Гирке болезнь. Гликогенозы Болезнь гирке биохимия
Болезнь Гирке ( Гликогеноз I типа , Дефект глюкозо-6-фосфаттранслоказы , Недостаточность глюкозо-6-фосфатазы )
Болезнь Гирке – это наследственная патология, которая обусловлена недостаточностью печеночных ферментов, трансформирующих глюкозо-6-фосфат в глюкозу. Протекает с нарушениями углеводного и липидного обмена, характеризуется гипогликемией, накоплением в печени и почках гликогена. Симптомы включают слабость, головные боли, судороги, тошноту, рвоту, артериальную гипотензию, одышку, повышение температуры тела, снижение свертываемости крови. Диагностика проводится лабораторными методами, включает биохимический анализ крови, провокационные тесты, биопсию и цитологическое исследование печени. Лечение основано на диетотерапии, обеспечивающей круглосуточное поступление глюкозы в кровоток.
МКБ-10
Общие сведения
Болезнь Гирке имеет ряд синонимичных названий: гликогеноз типа I, недостаточность глюкозо-6-фосфатазы, дефект глюкозо-6-фосфаттранслоказы. Эпонимическое название связано с фамилией врача из Германии Эдгара фон Гирке, исследования которого в 1929 году привели к открытию синтеза гликогена I типа. Он подробно описал биохимические изменения и клиническую картину гликогеноза I. Ферментный дефект был обнаружен в 1952 году. Распространенность синдрома невелика ‒ 1:200 тысяч новорожденных. При этом заболевание является наиболее частым среди гликогенозов. Частота одинакова среди представителей мужского и женского пола, диагноз устанавливается в возрасте до 1 года, как правило, до 3-5 месяцев.
Причины
Заболевание относится к наследственным метаболическим патологиям, передается от поколения к поколению по аутосомно-рецессивному механизму. Причина болезни – мутации генов, которые кодируют печеночный фермент, запускающий процесс превращения гликогена в свободную глюкозу. При изменении гена G6PC нарушено производство глюкозо-6-фосфатазы, при изменении в структуре гена SLC37A4 – синтез глюкозо-6-фосфаттранслоказы.
Для развития гликогенеза необходимо, чтобы ребенок получил мутировавший ген от матери и от отца. Патология развивается при наличии пары измененных рецессивных генов, а если присутствует только один дефектный ген, ребенок остается здоровым, но является носителем заболевания и способен передать его своим будущим детям. Таким образом, вероятность болезни у новорожденного, оба родителя которого – носители мутации, составляет 25%.
Патогенез
При болезни Гирке клетки печени оказываются неспособными производить ферменты, обусловливающие процессы гликогенолиза (расщепления гликогена до глюкозы) и глюконеогенеза (образования глюкозы из неуглеводных соединений). Преобразование поступающей с пищей глюкозы в гликоген, а также его депонирование в печеночной и других тканях остается сохранным. Гликоген накапливается, но не растрачивается на нужды организма, печень увеличивается. В перерывах между приемами пищи развивается гипогликемия из-за невозможности использования энергетических запасов.
Устойчивая гипогликемия провоцирует постоянное усиленное производство глюкагона, являющегося стимулятором гликогенолиза. Этот процесс прерывается недостатком ферментов, реализуются лишь его начальные стадии, и гипогликемия сохраняется. В клетках повышается содержание глюкозо-6-фосфата – промежуточного продукта гликогенолиза. Благодаря обратной связи гликоген остается в почках и печени, это проявляется структурными изменениями органов и увеличением их размеров. Формируется почечная и печеночная недостаточность. В кровотоке повышается концентрация молочной кислоты, возникает ацидоз.
Классификация
По характеру течения болезнь Гирке бывает острой и хронической. Острая форма чаще развивается на первом году жизни ребенка, проявляется выраженной симптоматикой: рвотой, мышечными судорогами, поверхностным частым дыханием, которое сопровождается чувством нехватки воздуха. Для хронической формы характерно постепенное прогрессирование почечной и печеночной недостаточности, отставание в росте, задержка пубертата. Согласно первоначальному ферментному дефекту выделяют два типа гликогеноза:
- ТипIa. При данном варианте нарушено производство глюкозной фосфатазы. Протекает в более мягкой форме, чем гликогеноз Ib. Составляет 80% случаев болезни.
- ТипIb. Определяется недостаточность фосфат-транслоказы. Развивается нейтропения, возрастает риск инфекционных осложнений.
Симптомы болезни Гирке
Симптоматика заболевания чаще всего разворачивается на 3-4 месяце жизни, реже – с периода новорожденности. Наиболее выражены проявления лактатацидоза и гипогликемии: малыши вялые, сонливые, плохо сосут грудь или соску бутылочки, часто плачут, подолгу не успокаиваются. Сон беспокойный, с частыми пробуждениями, режим дня и ночи не устанавливается, кровяное давление сниженное. Могут отмечаться непроизвольные мышечные подергивания на руках, ногах, веках. К 3 месяцам развиваются судороги. Все эти симптомы более заметны у детей, находящихся на искусственном вскармливании и получающих питание «по часам», а не по требованию. При частых кормлениях глюкоза поступает в организм практически непрерывно, гипогликемия слабовыраженная.
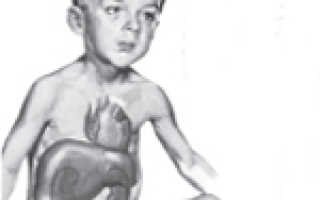
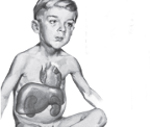
Изменяются особенности внешности ребенка: лицо становится «кукольным», округлым, щеки и живот увеличены, конечности относительно тонкие. По мере взросления определяется низкорослость, слабое физическое развитие, позднее половое созревание. Жировая клетчатка распределяется особым образом: ручки и ножки худые, кажутся еще более тонкими на фоне крупного лица и туловища. Большие размеры живота связаны как с накоплением подкожного жира, так и с патологическим увеличением печени.
Формируются ксантомы – очаговые жировые новообразования под кожей, расположенные на локтях, коленях, бедрах, ягодицах. Появляется одышка, температура нередко сохраняется на уровне субфебрильных значений 37-37,5˚C. Сниженная активность тромбоцитов проявляется носовыми кровотечениями, кровоподтеками. У некоторых детей периодически возникает диарея. Недостаточность функции почек развивается при длительном тяжелом течении болезни.
Осложнения
При неадекватном лечении, несоблюдении назначений врача приступы тяжелой гипогликемии негативно сказываются на работе высших отделов ЦНС, приводят к олигофрении. У части пациентов возникают серьезные поражения печени: к 20-30 годам развиваются аденомы, способные трансформироваться в злокачественную опухоль – карциному. К другим возможным осложнениям болезни Гирке относятся гиперурикемическая подагра, панкреатит, хроническая почечная недостаточность, амилоидоз, синдром Фанкони-Биккеля, гипокальциемия, дистальноканальцевый ацидоз.
Диагностика
Заболевание диагностируется в первые месяцы после рождения. Обследование детей проводят педиатры, эндокринологи, врачи-генетики. Подозрение на болезнь Гирке возникает при характерных клинических признаках, выявлении повышенного содержания липидных соединений и лактата в результатах анализа крови. Для подтверждения предполагаемого диагноза, дифференциации разных типов гликогенозов, исключения сифилиса, токсоплазмоза, цитомегалии, опухолей и стеатоза печени проводится ряд лабораторных исследований:
- Биохимическое исследование крови. При гликогенозе 1 типа количество глюкозы после 4 часов голода составляет не более 2,2 ммоль/л, после 5-7 часов без пищи – менее 1,10 ммоль/л. Определяется повышенный уровень молочной и мочевой кислот, триглицеридов, холестерина, ферментов АсАТ и АлАТ. Большое содержание жировых субстанций делает сыворотку мутной, молочного цвета.
- Лабораторные провокационные пробы. С целью дифференциации гликогенозов различных типов и определения дефекта ферментов выполняется замер уровня метаболитов и гормонов натощак, а затем после углеводной нагрузки. Для гликогеноза первого типа характерно, что после нагрузки глюкозой уровень лактата снижается, кривая имеет диабетоидную форму – высокий пик подъема сахара и медленное снижение.
- Глюкагоновая проба. Глюкагон вводят внутримышечно или внутривенно после 4-6-часового периода голода, отслеживают содержание лактата и глюкозы через определенные промежутки времени. При болезни Гирке глюкагон не способствует повышению концентрации глюкозы или повышает ее незначительно, высокие показатели лактата продолжают расти.
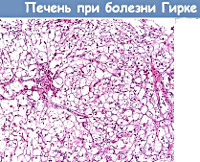
- Исследование биопсийного материала печени. Для точной диагностики ферментного дефекта исследуются цельные и разрушенные микросомы печени, определяется активность глюкозо-6-фосфатазы. Дифференциальным признаком гликогенеза Ia является снижение активности фермента в микросомах любого типа на 10%. При варианте болезни Ib он активен в поврежденных микросомах, а в целых отсутствует либо мало функционален. Также выявляется высокое содержание гликогена со стандартной структурой молекул.
Лечение болезни Гирке
Терапия нацелена на компенсацию гипогликемии, устранение вторичных метаболических расстройств. При раннем начале лечения – в периоды новорожденности и младенчества – зачастую достаточно использования методов диетотерапии. При взрослении ребенка нарастает гиперурикемия и гиперлипопротеидемия, ухудшается функционирование почек, печени, что требует проведения медикаментозного лечения, а иногда – хирургического вмешательства. Полный комплекс терапевтических мероприятий включает:
Диетотерапия
Чтобы компенсировать неспособность ферментативной системы к высвобождению глюкозы, используется система диетического питания, которая обеспечивает непрерывность поступления глюкозы на протяжении 24 часов. В медицинской практике используются два метода, позволяющих снабжать организм углеводами круглосуточно:
- Зондовое введение глюкозы. Вливание питательных растворов производится через назогастральный путь, реже – через гастростому. Режим процедур подбирается индивидуально. Большинству детей необходима установка зонда на ночь, так как это позволяет не нарушать сон.
- Употребление кукурузного крахмала. Днем детям дают высокоуглеводную пищу. Широко применяется регулярное кормление сырым кукурузным крахмалом. Глюкоза из него высвобождается постепенно, в течение нескольких часов, благодаря чему поддерживается нормогликемия.
Медикаментозная коррекцию метаболических нарушений
К зрелому возрасту становятся необходимы дополнительные меры для нормализации уровня мочевой кислоты и липидов (диетотерапии недостаточно). При гиперурикемии назначается ингибитор ксантиноксидазы аллопуринол, при гиперлипидемии – фибраты, статины. При первых признаках нарушения почечной функции применяются ингибиторы АПФ, цитраты.
Лечение нейтропении
При развитии гликогеноза подтипа Ib, сопровождающегося уменьшением количества гранулоцитов, показано введение колониестимулирующих факторов: гранулоцитарного и гранулоцитарно-макрофагального. Они препятствуют развитию нейтропении и снижают ее выраженность, в результате чего сокращается частота инфекционных заболеваний.
Лечение новообразований печени
Пациентам с объемными аденомами печени проводятся процедуры чрескожных инъекций этанола (ЧИЭ). При злокачественных опухолях возможна рекомендация ортотопической трансплантации печени.
Прогноз и профилактика
Пациенты, не получающие нужного лечения, погибают в период новорожденности, младенчества или раннего детства от выраженной гипогликемии и ацидоза. При адекватной терапии заболевание успешно компенсируется, с возрастом выраженность гипогликемии уменьшается. У взрослых усиливается активность другого фермента, способствующего образованию глюкозы – амило-1,6-глюкозидазы. В итоге соотношение скорости производства и утилизации глюкозы становится более уравновешенным. Профилактические меры должны проводиться при планировании и на начальных сроках беременности. Супружеским парам из групп риска необходима консультация генетика для расчета вероятности заболевания. Пренатальная диагностика позволяет выявить наличие патологии у плода.
Гликогенозы
Гликогенозы и агликогенозы (болезнь накопления гликогена, гликогеновая болезнь) представляют собой группу передающихся по наследству заболеваний. Характеризуются они различного рода нарушениями синтеза и/или расщепления гликогена (сложного углевода, который состоит из соединенных в цепочку молекул глюкозы), а также его избыточным отложением в тканях организма, преимущественно в печени и мышцах.
Нарушения метаболизма, сопровождающиеся избыточным накоплением в тканях гликогена или же характеризующиеся образованием в них патологически измененных разновидностей гликогена, носят название гликогенозы. И наоборот, нарушения, связанные с недостаточным содержанием гликогена в тканях и сопровождающиеся проявлениями судорожного синдрома, отставанием в умственном и физическом развитии, получили название агликогенозы.
Гликогенозы и агликогенозы обусловливаются ферментативной недостаточностью и встречаются у одного человека на 40-68 тысяч населения.
В зависимости от того, активность какого фермента нарушилась, в настоящее время принято выделять двенадцать типов гликогенозов. Все они, за исключением IV типа, наследуются по аутосомно-рецессивному типу.
Разновидности гликогенозов
Помимо классификации по номерам, заболевания, связанные с нарушением метаболизма гликогена, также принято делить по патогенетическому признаку на гликогенозы:
1. Печеночные гликогенозы, развиваются вследствие нарушения нормальных ферментативных реакций в печени. Характеризуются избыточным отложением гликогена в печени, чем провоцируется ее патологическое увеличение (гепатомегалия) с сопутствующими ему симптомами.
Наиболее распространенным среди заболеваний данного типа является болезнь Гирке (или гликогеноз I типа), сопровождающаяся нарушением депонирования гликогена в печени. При этом нарушения, затрагивающие процессы обмена гликогена, по сути вторичны, а их развитие провоцирует ферментативная недостаточность глюкозо-6-фосфатазы, которая наследуется по рецессивному типу и вследствие которой становится невозможным превращение глюкозо-6-фосфата в глюкозу.
У пациентов с болезнью Гирке отмечается низкий уровень содержания глюкозы в крови натощак. Для того чтобы в крови поддерживалось нормальное количество глюкозы, требуется практически непрерывное ее поступление вместе с пищей. Однако в реальных условиях, когда непрерывное поступление глюкозы является невозможным, здоровый человеческий организм запасает и при необходимости расходует гликоген, образующийся вследствие полимеризации глюкозы.
При болезни Гирке у пациентов способность организма синтезировать из глюкозы гликоген сохраняется, как и способность откладывать гликоген в тканях различных органов (главным образом печени), обратный же процесс в тех случаях, когда уровень глюкозы в крови снижается, при этом невозможен. Таким образом, преобразование глюкозы в гликоген, являющееся для здорового человека физиологически нормальным процессом, в таких условиях способно спровоцировать развитие дополнительных патологий. В особо тяжелых ситуациях, гипогликемия вызывает сильные судороги, а вследствие хронически подавляемой выработки инсулина происходит задержка роста.
Гликогеноз III типа также принадлежит к категории печеночных гликогеновых заболеваний. Его другие названия – болезнь Форбса, болезнь Кори или лимитдекстриноз. Главным его признаком является скопление избыточного количества аномального гликогена в тканях печени, в миокарде и в скелетных мышцах. Заболевание наследуется по аутосомно-рецессивному типу и провоцируется отсутствием фермента амило-1,6-глюкозидазы, являющегося катализатором распада 1-6 связей в молекуле гликогена.
Клиническая картина по своим проявлениям сходна с проявлениями гликогенозом I типа, однако выражена не так ярко. Дефект фермента выявляется в лейкоцитах крови, наличии аномального гликогена в эритроцитах. При болезни Кори молекулы гликогена отличаются наличием очень коротких наружных ветвей (в отличие от гликогеноза IV типа, характеризующегося недостаточностью ветвящего фермента и отложением гликогена с аномально длинными цепями в молекуле). Однако, несмотря на то, что при гликогенозе IV типа (болезнь Андерсена, спровоцированная дефектом фермента амило-(1,4-1,6)-трансглюкозилазы) в тканях печени откладывается значительно меньше гликогена, чем при гликогенозе III типа, ее течение является менее доброкачественным и несет угрозу жизни ребенка (гликогеновая болезнь III типа имеет доброкачественное течение и не является угрозой для жизни). Это связано с тем, что при IV типе гликогеноза организм реагирует на присутствие в нем аномального гликогена развитием цирроза печени, что, в свою очередь, приводит к летальному исходу.

Гликогеноз VI типа представляет собой достаточно редкое заболевание. Оно также называется болезнью Херса и обусловливается наличием дефектов фосфорилазы в печени. Патология сопровождается выраженной гепатомегалией при отсутствии каких-либо нарушений функций печени и задержки роста. При этом в тканях печени депонируется гликоген, структура которого является полностью нормальной. Симптоматически этот вид гликогеноза сходный с гликогеновой болезнью I типа, однако его проявления выражены не настолько сильно.
2. Мышечные гликогенозы характеризуются тем, что нарушения, которые они вызывают, затрагивают энергоснабжение скелетных мышц. Этот тип заболеваний выявляется, как правило, при физических нагрузках и проявляется в виде:
- Миальгии (выраженной мышечной боли);
- Миоглобинурии;
- Судорожного синдрома;
- Повышенной слабости;
- Ферментемии мышечными энзимами.
К этой группе заболеваний относятся гликогеноз V типа (или болезнь Мак-Ардля), гликогеноз VII типа, а также непронумерованные в соответствии с классификацией Кори дефицит мышечной фосфоглицеромутазы и дефект М-субъединицы лактатдегидрогеназы (ЛДГ).
Болезнь Мак-Ардля считается наиболее редко встречающейся патологией. Она наследуется по аутосомно-рецессивному типу и поражает исключительно скелетные мышцы, в которых наблюдается полное отсутствие фосфорилазы.
Характерной особенностью гликогеноза VII типа является сниженное содержание фосфофруктокиназы в скелетной мускулатуре (порядка 13% от ее нормального количества в здоровом организме). Клиническая картина данного гликогеноза сходна с проявлениями болезни Мак-Ардля, протекающей в нетяжелой форме, при этом пациенты способны выдерживать умеренную физическую активность.
3. Гликогенозы смешанного типа (генерализованные) затрагивают как печень и скелетную мускулатуру, так и прочие органы. Их симптомы гораздо более разнообразны, нежели симптомы гликогеновой болезни печеночного или мышечного типа, а течение более прогрессирующее.
Основа лечения гликогенозов и агликогенозов
Лечение гликогенозов и агликогенозов зависит исключительно от типа заболевания. Но в любом случае его главной целью остается недопущение развития гипогликемии.
Гирке болезнь: причины, симптомы, лечение
Гликогеноз типа 1 впервые был описан в 1929 году Гирке. Болезнь встречается в одном случае из двухсот тысяч новорожденных. Патология поражает одинаково как мальчиков, так и девочек. Далее рассмотрим, как проявляется болезнь Гирке, что это, какая терапия применяется.

Общие сведения
Несмотря на сравнительно раннее обнаружение, только в 1952 году Кори был установлен ферментный дефект. Наследование патологии аутосомно-рецессивное. Синдром Гирке – болезнь, на фоне которой клетки печени и извитых канальцев почек заполняются гликогеном. Однако эти резервы оказываются недоступными. На это указывает гипогликемия и отсутствие увеличение в крови концентрации глюкозы в ответ на глюкагон и адреналин. Синдром Гирке – болезнь, сопровождающаяся гиперлипемией и кетозом. Данные признаки являются характерными для состояния организма при дефиците углеводов. При этом в печени, кишечных тканях, почках отмечается низкая активность глюкозо-6-фосфатазы (либо она отсутствует совершенно).
Ход патологии
Как развивается синдром Гирке? Болезнь обуславливается дефектами в ферментной системе печени. Она превращает в глюкозу глюкозо-6-фосфат. При дефектах нарушается как глюконеогенез, так и гликогенолиз. Это, в свою очередь, провоцирует гипертриглицеридемии и гиперурикемии, лактацидоз. В печени происходит скопление гликогена.
Болезнь Гирке: биохимия
В ферментной системе, которая трансформирует в глюкозу глюкозо-6-фосфат, кроме него самого, присутствует еще не менее четырех субъединиц. К ним, в частности, относят регуляторный Са2(+)-связывающее белковое соединение, транслоказы (белки-переносчики). В системе содержится Т3, Т2, Т1, обеспечивающие трансформацию глюкозы, фосфата и глюкозо-6-фосфата сквозь мембрану ретикулума эндоплазмы. Существуют определенные сходства у типов, которые имеет болезнь Гирке. Клиника гликогеноза Ib и Ia аналогична, в связи с этим для подтверждения диагноза и точного установления ферментного дефекта проводится биопсия печени. Также исследуется активность глюкозо-6-фосфатазы. Разница в клинических проявлениях между гликогенозом типа Ib и Ia состоит в том, что при первом отмечается преходящая либо постоянная нейтропения. В особо тяжелых случаях начинает развиваться агранулоцитоз. Нейтропению сопровождает дисфункция моноцитов и нейтрофилов. В связи с этим увеличивается вероятность кандидоза и стафилококковых инфекций. У отдельных пациентов появляется воспаление в кишечнике, схожее с болезнью Крона.
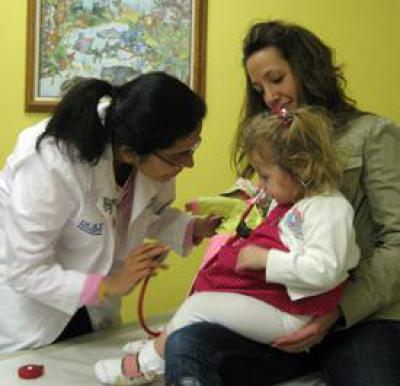
Признаки патологии
В первую очередь следует сказать, что у новорожденных, детей грудного и старшего возраста по-разному проявляется болезнь Гирке. Симптомы проявляются в виде гипогликемии голодания. Однако в большинстве случаев патология протекает бессимптомно. Это связано с тем, что грудные дети достаточно часто получают питание и оптимальное количество глюкозы. Болезнь Гирке (фото заболевших можно найти в медицинских справочниках) нередко диагностируется после рождения спустя несколько месяцев. У ребенка при этом выявляется гепатомегалия и увеличение живота. Субфебрильная температура и одышка без признаков инфекции также могут сопровождать болезнь Гирке. Причины последней – лактацидоз вследствие недостаточной выработки глюкозы и гипогликемия. С течением времени интервалы между кормлениями увеличиваются и появляется продолжительный ночной сон. При этом отмечаются симптомы гипогликемии. Ее продолжительность и тяжесть начинает постепенно увеличиваться, что, в свою очередь, приводит к метаболическим расстройствам системного типа.
Последствия
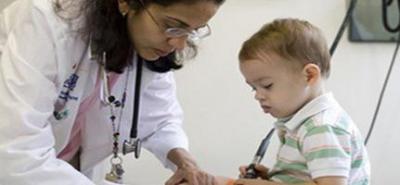
При отсутствии лечения отмечаются изменения во внешности ребенка. В частности, характерными являются мышечная и скелетная гипотрофия, замедление физического развития и роста. Наблюдаются также жировые отложения под кожей. Ребенок начинает походить на больного, у которого синдром Кушинга. При этом не отмечается нарушений в развитии социальных и познавательных навыков, если при повторных гипогликемических приступах не был поврежден головной мозг. Если сохраняется гипогликемия голодания и ребенок не получает необходимого объема углеводов, задержка в физическом развитии и росте становится четко выраженной. В некоторых случаях дети с гипогликенозом I типа умирают вследствие легочной гипертензии. При нарушении функции тромбоцитов наблюдаются повторные носовые кровотечения или кровоточивость после проведения стоматологического или иного хирургического вмешательства.
Аденомы печени
Они возникают у многих пациентов по разным причинам. Как правило, проявляются они в возрасте от 10 до 30 лет. Они могут малигнизироваться, возможны кровоизлияния в аденому. Эти образования на сцинтиграммах представлены в виде участков пониженного скопления изотопа. Для выявления аденом используется ультразвуковое исследование. В случае подозрения на злокачественное новообразование применяют более информативные МРТ и КТ. Они позволяют проследить трансформацию четкого ограниченного формирования небольшого размера в более крупное с достаточно размытыми краями. При этом рекомендовано периодическое измерение в сыворотке уровня альфа-фетопротеина (маркер рака клеток печени).
Диагностика: обязательные исследования
Пациентам измеряют уровни мочевой кислоты, лактата, глюкозы, активность печеночных ферментов натощак. У грудничков и новорожденных детей концентрация глюкозы в крови спустя 3-4-часовое голодание снижается до 2,2 ммоль/литр и более; при продолжительности больше четырех часов концентрация практически всегда меньше 1,1 ммоль/литр. Гипогликемию сопровождает значительное повышение содержания лактата и метаболический ацидоз. Сыворотка, как правило, мутная либо похожа на молоко вследствие очень высокой концентрации триглицеридов и умеренно увеличенного уровня холестерина. Наблюдаются также усиление активности АлАТ (аланинаминотрансферазы) и АсАТ (аспартаминотрансферазы), гиперурикемия.
Провокационные пробы
Для дифференциации типа I от прочих гликогенозов и точного определения ферментного дефекта у детей грудного и старшего возраста измеряется уровень метаболитов (жирных свободных кислот, глюкозы, мочевой кислоты, лактата, кетоновых тел), гормонов (СТГ (соматотропного гормона), кортизола, адреналина, глюкагона, инсулина) после глюкозы и натощак. Исследование осуществляется по определенной схеме. Ребенок получает глюкозу (1,75 г/кг) внутрь. Далее каждые 1-2 часа производится забор крови. Концентрация глюкозы быстро измеряется. Последний анализ берут не позднее шести часов после приема глюкозы или когда ее содержание уменьшилось до 2,2 ммоль/литр. Также проводится провокационная проба с глюкагоном.
Специальные исследования
В ходе них проводится биопсия печени. Также исследуется гликоген: его содержание значительно увеличено, но структура в пределах нормы. Осуществляются измерения активности глюкозо-6-фосфатазы в разрушенных и цельных микросомах печени. Их разрушают при помощи повторного замораживания и оттаивания биопата. На фоне гликогеноза типа Ia активность не определяется ни в разрушенных, ни в цельных микросомах, при типе Ib – в первых она нормальная, а во вторых существенно снижена либо отсутствует.
Болезнь Гирке: лечение
При гликогенозе типа I метаболические нарушения, связанные с недостаточной выработкой глюкозы, появляются уже после еды спустя несколько часов. При продолжительном голодании расстройства значительно усиливаются. В связи с этим лечение патологии сводится к учащению кормления ребенка. Цель терапии состоит в предупреждении падения содержания глюкозы ниже 4,2 ммоль/литр. Это пороговый уровень, при котором стимулируется секреция контрисулярных гормонов. Если ребенок получает своевременно достаточный объем глюкозы, отмечается уменьшение размеров печени. Лабораторные показатели при этом приближаются к норме, а психомоторное развитие и рост стабилизируются, исчезает кровоточивость.
Что такое гликогеновые болезни?
Гликогеновые болезни – это наследственные заболевания, обусловленные недостаточностью каких-либо ферментов, отвечающих за метаболизм гликогена. Могут быть нарушены обе стороны обмена: как синтез гликогена, так и его распад. Средняя частота встречаемости составляет 1:40000.
Гликогенозы
Синдром гликогеноза возникает в результате дефекта фермента синтеза или мобилизации гликогена, что приводит к накоплению или изменению структуры гликогена в разных тканях, чаще в печени и мышцах. Следует отметить, что при гликогенозах количество гликогена не всегда изменено, изменения могут быть только в структуре его молекулы.
Всего известно 12 типов гликогенозов. По патогенетическому признаку гликогенозы делят:
- печеночные – 0, I, III, IV, VI, VIII, IX, Х, ХI типов,
- мышечные – V и VII типов,
- смешанные – II типа.
Печеночные гликогенозы
Самый частый гликогеноз I типа или болезнь фон Гирке (частота 1 : 50000-100000 новорожденных) обусловлен аутосомно-рецессивным дефектом глюкозо-6-фосфатазы . Из-за того, что этот фермент есть только в печени и почках, преимущественно страдают эти органы, и болезнь носит еще одно название – гепаторенальный гликогеноз. Даже у новорожденных детей наблюдаются гепатомегалия и нефромегалия, обусловленные накоплением гликогена не только в цитоплазме, но и в ядрах клеток. Кроме этого, активируется синтез липидов с возникновением стеатоза печени. Так как фермент необходим для дефосфорилирования глюкозо-6-фосфата с последующим выходом глюкозы в кровь, у больных отмечается гипогликемия и, как следствие, ацетонемия, метаболический ацидоз, ацетонурия.
Гликогеноз III типа или болезнь Форбса-Кори или лимит-декстриноз – это аутосомно-рецессивный дефект амило-α1,6-глюкозидазы, “деветвящего” фермента, гидролизующего α1,6-гликозидную связь. Болезнь имеет более доброкачественное течение, и частота ее составляет примерно 25% от всех гликогенозов. Для больных характерна гепатомегалия, умеренная задержка физического развития, в подростковом возрасте возможна небольшая миопатия.
При гликогенозе IV типа (болезнь Андерсена, 1% всех гликогенозов), связанного с дефектом ветвящего фермента, образуется гликоген с малым количеством ветвлений, что резко уменьшает скорость гликогенолиза.
Гликогеноз VI типа (болезнь Херса, 25% всех гликогенозов), связан с дефицитом печеночной фосфорилазы гликогена. При этом отсутствует мобилизация гликогена, развивается гепатомегалия и гипогликемия.
Мышечные гликогенозы
Для этой группы гликогенозов характерны изменения ферментов мышечной ткани. Это приводит к нарушению энергообеспечения мышц при физической нагрузке, к болям в мышцах, судорогам.
Гликогеноз V типа (болезнь Мак-Ардля) – отсутствие мышечной фосфорилазы . При тяжелой мышечной нагрузке возникают судороги, миоглобинурия, хотя легкая работа не вызывает каких-либо проблем.
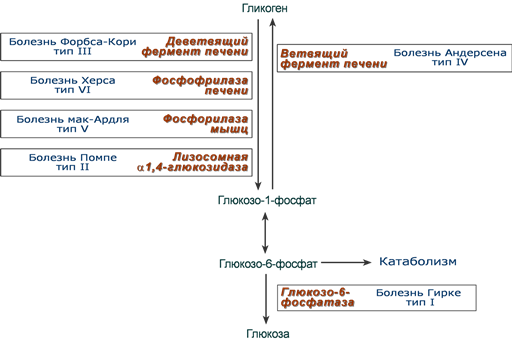
Схематичное расположение дефектных ферментов при различных гликогенозах
Смешанные гликогенозы
Эти заболевания касаются и печени, и мышц, и других органов.
Гликогеноз II типа (болезнь Помпе, 10% всех гликогенозов) – поражаются все гликогенсодержащие клетки из-за отсутствия лизосомальной (кислой) α-1,4-глюкозидазы , поэтому данная болезнь относится к лизосомным болезням накопления. Происходит накопление гликогена в лизосомах и в цитоплазме. Заболевание составляет почти 10% всех гликогенозов и является наиболее злокачественным. Больные при отсутствии лечения умирают в раннем возрасте из-за кардиомегалии и тяжелой сердечной недостаточности.
Агликогенозы
Агликогенозы – состояния, связанные с отсутствием гликогена. В качестве примера агликогеноза можно привести наследственный аутосомно-рецессивный дефицит гликоген-синтазы. Симптомами является резкая гипогликемия натощак, особенно утром, появляется рвота, судороги, потеря сознания. В результате гипогликемии наблюдается задержка психомоторного развития, умственная отсталость. Болезнь несмертельна при адекватном лечении (частое кормление), хотя и опасна.