Признаки единичных мелких очагов лейкодистрофии. Что за болезнь лейкодистрофия Пелицеуса — Мерцбахера? Общие проявления для всех видов
Лейкодистрофия
Лейкодистрофия — нейродегенеративное заболевание, обусловленное наследственным нарушением обмена веществ с накоплением в головном и спинном мозге метаболитов, провоцирующих разрушение миелина. Манифестирует в основном в детском возрасте задержкой психомоторного развития, двигательными расстройствами, поражением зрительных и слуховых нервов, гидроцефалией, эпилептическими приступами. Диагностируется лейкодистрофия по данным неврологического статуса, анамнеза, генетических исследований, МРТ или КТ картины головного мозга, биохимических анализов. Лечение симптоматическое. При раннем выявлении и медленном прогрессировании возможна трансплантация пуповинной крови или костного мозга.
МКБ-10
Общие сведения
Лейкодистрофия получила свое название в связи с поражением белого вещества мозга (с греческого leukos — белый). Различают около 60 разновидностей лейкодистрофии, определяющихся видом генной аномалии и возрастом манифестации клинических проявлений. Наряду с отдельными воспалительными поражениями ЦНС (например, лейкоэнцефалитом Шильдера) лейкодистрофия относится к синдрому диффузного склероза мозга. При этом доминирующее поражение миелина сближает ее с демиелинизирующими заболеваниями (рассеянным склерозом, РЭМ и пр.), а отдельные формы можно отнести к липидозам.
К основным формам лейкодистрофии относятся метахроматическая, суданофильная, глобоидно-клеточная, дегенерация Ван-Богарта-Бертрана, болезнь Александера, вариант Галлервордена-Шпатца. Наиболее распространены первые 3 вида лейкодистрофии. Их встречаемость колеблется от 0,4 до 1 случая на 100 тыс. новорожденных. Ряд форм лейкодистрофии являются настолько редкими, что в мировой литературе по неврологии описано всего несколько сотен их клинических наблюдений. В зависимости от возрастного периода, в котором дебютирует лейкодистрофия, каждая ее форма может подразделяться на инфантильный, поздний инфантильный, ювенильный и взрослый вариант.
Причины возникновения лейкодистрофии
В своей основе каждая лейкодистрофия имеет генетическую аномалию определенного фермента. Вид аномалии и локализация генной мутации пока установлены лишь для наиболее встречающихся форм патологии. В большинстве случаев лейкодистрофия имеет аутосомно-рецессивный путь наследственной передачи, однако отдельные ее формы могут наследоваться сцеплено с полом. Кроме того, не одиноки случаи спонтанных мутаций. Генетически детерминированный энзимный дефект ведет к обменным нарушениям (чаще в метаболизме липидов) с отложением определенного метаболита в нервных структурах и отдельных соматических органах, в первую очередь в печени и почках.
Следствием метаболической аномалии является разрушение миелина оболочек нервных стволов и проводящих путей, гибель нейронов с замещением их разрастающейся глиальной тканью. Морфологически лейкодистрофия характеризуется диффузными и симметрично расположенными в полушариях головного мозга зонами гибели миелина, скоплением продуктов миелинового распада, усиленной пролиферацией глии. В отдельных нозологических вариантах лейкодистрофия имеет специфическую морфологическую картину — метахроматическое или суданофильное окрашивание продуктов миелинового распада, скопление в зонах демиелинизации глобоидных клеток и т. п.
Симптомы лейкодистрофии
В большинстве случаев лейкодистрофия дебютирует в раннем детском возрасте. Новорожденные, как правило, выглядят здоровыми. Определенный период они нормально развиваются, а затем постепенно возникают различные неврологические симптомы, отличающиеся неуклонным прогрессированием. Скорость нарастания симптомов тем выше, чем раньше манифестировала лейкодистрофия. Ведущими проявлениями выступают прогрессирующая олигофрения, ухудшение зрения, тугоухость, эписиндром, спастические парезы. Первыми симптомами лейкодистрофии могут быть атаксия, мышечно-тонические расстройства (гипо- или гипертонус, мышечные подергивания), экстрапирамидные проявления, изменения поведения. Затем возникают эпиприступы, бульбарные проявления, снижается слух и зрение, отмечается интеллектуальное снижение с постепенной утратой ранее приобретенных навыков. Сенсорные расстройства не характерны. На поздних этапах развития болезни наблюдаются параличи, выраженная олигофрения, грубое расстройство глотания, амавроз, глухота. В терминальной фазе обычно отмечается децеребрационная ригидность.
Виды лейкодистрофии
Метахроматическая лейкодистрофия в зависимости от манифестации имеет 4 варианта. Врожденный вариант дебютирует в первые 1-3 мес. жизни задержкой развития и судорожным синдромом; дети не достигают возраста 1 года. Позднедетский вариант метахроматической лейкодистрофии начинается в период от 1 до 3 лет с мышечной гипотонии и слабости, атаксии, задержки психического развития (ЗПР). Затем формируется спастическая тетраплегия, афазия, псевдобульбарный синдром. В редких случаях пациенты доживают до 10-летнего возраста. Ювенильный вариант манифестирует в 4-6 лет и длится в среднем 7 лет. Взрослый вариант дебютирует в третьей декаде жизни, иногда позднее, продолжительность жизни пациентов от начала клиники варьирует в пределах 10-20 лет.
Суданофильная лейкодистрофия наследуется сцеплено с Х-хромосомой и имеет несколько разновидностей. Лейкодистрофия Пелицеуса-Мерцбахера может стартовать на 1-ом году жизни или в 3-4 года. Первым признаком является крупноразмашистый нистагм, позже возникает ЗПР, мозжечковая атаксия, гиперкинезы, парезы. Наибольшее прогрессирование происходит в возрасте до 10 лет, затем заболевание принимает замедленное течение с длительными ремиссиями. Пациенты могут жить до зрелого возраста. Адренолейкодистрофия — вариант, при котором лейкодистрофия сочетается с надпочечниковой недостаточностью. Характеризуется прогрессирующим течением с летальным исходом спустя 6-8 лет от начала клиники.
Глобоидно-клеточная лейкодистрофия (болезнь Краббе) — липоидоз с накоплением в очагах демиелинизации галактоцереброзида и образованием больших округлых глобоидных клеток. Раннедетский вариант развивается в первом полугодии жизни с гипервозбудимости и периодической гипертермии, задерживается психомоторное развитие, нарастает тонус мышц, затем развивается спастический тетрапарез, олигофрения, эписиндром, возможен опистотонус. В годовалом возрасте наступает летальный исход. Позднедетский вариант более редкий, манифестирует ухудшением зрения.
Спонгиозная дегенерация Ван-Богарта-Бертрана характеризуется эписиндромом, гиперсомнией, выраженной гидроцефалией с увеличением размеров головы, вызывающей амавроз атрофией зрительных нервов. Резкая внутричерепная гипертензия приводит к расхождению черепных швов, регистрируемому при рентгенографии черепа. Пациенты с этой формой лейкодистрофии погибают до 3-летнего возраста.
Болезнь Александера (лейкодистрофия с волокнистой формацией) обусловлена мутацией гена, ответственного за синтез GFAP белка. В результате происходит накопление в клетках глии аномального GFAP белка, содержащего волокна Розенталя. Неонатальный вариант имеет тяжелое течение с летальным исходом к концу 1-го года. Инфантильный вариант встречается примерно в половине случаев, проявляется в первые 1-2 года жизни ЗПР, затем присоединяются спастические парезы, атаксия, гидроцефалия. Дети погибают спустя несколько лет. Ювенильная лейкодистрофия Александера дебютирует в период от 4-х до 10-летнего возраста, протекает с преимущественно стволовой симптоматикой. Продолжительность жизни колеблется в пределах 10-30 лет. Взрослый вариант отличается поздней манифестацией и относительно медленным течением в пределах 10 и более лет.
Лейкодистрофия Галлервордена-Шпатца чаще всего стартует в 10-летнем возрасте. Проявляется дисфункцией стриопаллидарной системы, затем на фоне гиперкинезов прогрессирует тетрапарез, развивается гемералопия и пигментный ретинит, наблюдается снижение интеллекта, возникают эпиприступы.
Диагностика лейкодистрофии
Диагностический поиск требует привлечения ряда специалистов: невролога, педиатра, медицинского генетика, для диагностики расстройств зрения и слуха — отоларинголога и офтальмолога. Важное значение имеет изучение анамнеза болезни (возраст и симптомы дебюта, последовательность развития клиники) и семейного анамнеза (наличие лейкодистрофии у родственников). Нейросонография через родничок и эхо-энцефалография у пациентов более старшего возраста, как правило, выявляет повышение интракраниального давления. Лейкодистрофия сопровождается существенным увеличением концентрации белка, обусловленным разрушением церебральных клеток, что определяется при исследовании цереброспинальной жидкости.
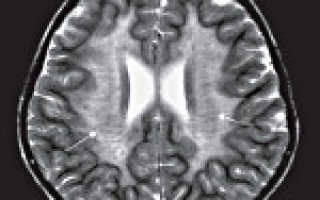
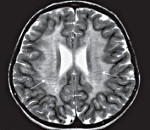
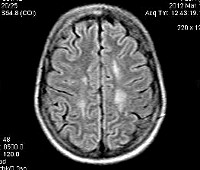
С целью диагностики вида метаболической аномалии проводится целый ряд биохимических тестов с определением уровня ферментов и накапливающихся метаболитов. Очаги демиелинизации хорошо визуализируются при помощи МРТ, могут быть обнаружены и на КТ головного мозга. Обычно демиелинизация видна на МРТ головного мозга еще до клинической манифестации лейкодистрофии. Благодаря развитию генетики, лейкодистрофия имеет разработанную ДНК-диагностику, а отдельные ее формы (метахроматическая, адренолейкодистрофия, глобоидно-клеточная) — возможность пренатального диагностирования.
Лечение лейкодистрофии
На сегодняшний день лейкодистрофия не имеет эффективных способов терапии, позволяющих купировать прогрессирование симптомов. Проводится симптоматическое лечение — в основном дегидратационная и антиконвульсантная терапия. Единственным методом, способным увеличить продолжительность жизни пациентов с лейкодистрофией и улучшить качество их жизни, является трансплантация пуповинной крови или пересадка костного мозга. Трансплантация приводит к нормализации метаболизма. Однако этот процесс занимает длительное время (от 12 до 24 мес.), в течение которого продолжается прогрессирование лейкодистрофии. Поэтому зачастую тяжелая инвалидизация или гибель пациента наступает даже после успешной трансплантации.
Следует подчеркнуть, что трансплантация никак не влияет на уже развившийся неврологический дефицит, она лишь позволяет приостановить его дальнейшее прогрессирование. В связи с тем, что эффект такого лечения наступает спустя 1-2 года, оно целесообразно в случае ранней доклинической диагностики лейкодистрофии (при соответствующей настороженности родителей рожденного ребенка в связи с наличием подобной патологии в семье) или при медленно прогрессирующем варианте течения. Кроме того, необходимо учитывать, что трансплантация связана с риском ряда серьезных осложнений, таких как отторжение, реакция «трансплантат против хозяина», развитие инфекций.
Лейкодистрофия

- Большая голова
- Гипертонус мышц
- Задержка в психомоторном развитии
- Изменение поведения
- Нарушение глотания
- Нарушение координации движения
- Нарушение психического развития
- Нарушение речи
- Непроизвольные подергивания мышц
- Отставание в умственном развитии
- Повышенная нервная возбудимость
- Повышенное внутричерепное давление
- Слабость
- Снижение зрения
- Снижение интеллекта
- Снижение мышечного тонуса
- Снижение слуха
- Судороги
- Частичный паралич
- Эпилептические припадки
Лейкодистрофия – это патология нейродегенеративного генеза, которой существует более шестидесяти разновидностей. Для болезни характерно нарушение обмена веществ, что приводит к скоплению в головном или спинном мозге специфических компонентов, разрушающих такое вещество, как миелин.
Причины возникновения недуга заключаются в генных мутациях, но некоторые формы могут наследоваться от одного из родителей. Помимо этого, зафиксированы случаи спонтанных мутаций.
Симптоматика болезни будет отличаться в зависимости от того, в какой форме протекает недуг. Наиболее часто выражаются признаки олигофрении, снижение остроты слуха или зрения, а также пониженный или повышенный тонус мышц.
Поставить правильный диагноз можно на основании генетических исследований и инструментальных обследований пациента. Лечение носит симптоматический характер, но при раннем выявлении недуга для сохранения жизни ребёнка могут потребоваться специфические хирургические вмешательства.
Этиология
Лейкодистрофия, или прогрессирующий склероз мозга, получила своё название оттого, что в патологический процесс вовлекается белое вещество этого органа. На сегодняшний день известно большое количество форм болезни, отличающихся видом генетической мутации и возрастной категорией, в которой происходит манифестация симптомов.
Наиболее частые типы болезни, например, метахроматическая лейкодистрофия, диагностируются у одного младенца на сто тысяч новорождённых. Однако есть такие виды патологии, которых зарегистрировано не больше нескольких сотен.
Основной причиной любого вида недуга является генетическая аномалия того или иного фермента. Разновидности и локализация мутировавших генов установлены только для наиболее частых форм патологии.
Зачастую лейкодистрофия характеризуется аутосомно-рецессивным путём наследования, но некоторые виды могут передаваться сугубо по половой принадлежности, т. е. от матери к дочери или от отца к сыну.
Генетически обусловленный дефект наиболее часто приводит к нарушению обменных процессов, что чревато скоплением того или иного вещества в организме. В основном поражаются следующие органы:
Следствием обменного нарушения является:
- разрушение миелина в оболочках нервных стволов;
- гибель или атрофия нейронов;
- замещение погибших нейронов глиальной тканью, которая постоянно разрастается.
По морфологическим признакам лейкодистрофия характеризуется:
- диффузным или симметричным расположением областей гибели миелина в обоих полушариях головного мозга;
- скоплением большого количества продуктов, выделяющихся после распада миелина;
- усиленным разрастанием глии.
Для всех групп болезни характерно развитие в раннем детском возрасте, ещё до того, как ребёнок пойдёт в школу.
Классификация
В зависимости от того, в какой возрастной категории произойдёт манифестация подобной патологии, она имеет следующие формы:
- инфантильную – симптоматика начинает выражаться в промежутке от трёх до шести первых месяцев жизни;
- позднюю инфантильную – является таковой, если поставят диагноз в периоде, который начинается с полугода и заканчивается в полтора года.
- ювенильную или типичную детскую – болезнь манифестирует в возрасте от трёх до десяти лет;
- взрослую – отличается тем, что первые симптомы могут возникнуть начиная с шестнадцати лет.
Симптоматика
Зачастую формы лейкодистрофии выражаются в детском возрасте, при этом новорождённые в подавляющем большинстве случаев выглядят совершенно здоровыми. Определённый промежуток времени у ребёнка наблюдается нормальное развитие, которое соответствует его возрастной категории. Однако постепенно будут проявляться различные признаки неврологического характера, склонные к постоянному прогрессированию.
В зависимости от того чем раньше произошла манифестация, тем быстрее будет прогрессировать патология. Несмотря на то что клинические проявления зачастую зависят от вида лейкодистрофии, начальные признаки будут примерно одинаковыми.
Таким образом, группа первых симптомов включает в себя:
- олигофрению;
- ухудшение зрительной функции;
- симптоматическую эпилепсию;
- стойкое ослабление слуха;
- парезы спастического характера;
- гипотонус или гипертонус мышц;
- нарушение координации движений;
- непроизвольные мышечные подёргивания;
- резкие изменения в поведении;
- отставание в умственном развитии – более того, дети со временем теряют уже приобретённые навыки;
- нарушение процесса глотания;
- параличи.
Для метахроматической дистрофии характерны такие признаки:
- понижение мышечного тонуса, что приводит к постоянной слабости ребёнка;
- атаксия;
- нарушение психического развития;
- формирование спастической тетраплегии;
- частичная или полная утрата способности пользоваться собственной речью;
- развитие псевдобульбарного синдрома.
Тяжёлая клиническая картина приводит к тому, что пациенты с такой формой патологии довольно редко доживают до десятилетнего возраста. Если манифестация произошла у взрослого, то период с момента появления первых симптомов и до летального исхода составляет примерно двадцать лет.
Суданофильная разновидность патологии делится на несколько видов. Первый – лейкодистрофия Пелицеуса-Мерцбахера. В подавляющем большинстве случаев развивается либо на первом году жизни, либо в три года. Среди симптомов стоит выделить:
- нистагм;
- задержку психического развития;
- мозжечковую атаксию;
- гиперкинез;
- парез конечностей.
Примечательно то, что после того, как пациенту исполняется десять лет, прогрессирование болезни замедляется, что даёт возможность человеку дожить до зрелого возраста.

Второй тип – адренолейкодистрофия, помимо вышеуказанной симптоматики будут присутствовать проявления, характерные для такого недуга, как надпочечниковая недостаточность. Отличается от первой формы тем, что быстро прогрессирует и приводит к смерти пациента спустя восемь лет после начала манифестации.
Лейкодистрофия Краббе или глобоидно-клеточная болезнь развивается в первые шесть месяцев жизни малыша и выражается в:
- повышенной возбудимости;
- задержке психомоторного развития;
- нарастании тонуса мышц;
- развитии спастического тетрапареза;
- олигофрении;
- судорожных припадках.
Такие симптомы приводят к тому, что малыш умирает, не доживая до одного года.
Спонгиозная лейкодистрофия или болезнь Канавана-Бертрана в своём симптомокомплексе имеет:
- эписиндром;
- гиперсомнию;
- ярко выраженную гидроцефалию;
- увеличенные размеры головы по отношению ко всему туловищу;
- повышенное внутричерепное давление;
- атрофию зрительных нервов.
Дети с такой формой недуга зачастую погибают в трёхлетнем возрасте.
Болезнь Александера – ещё одна разновидность патологии, для которой характерны:
- гидроцефалия;
- спастические парезы;
- задержка психомоторного развития;
- атаксия.
Примечательно то, что чем позже манифестирует болезнь, тем дольше проживёт человек. Максимальная продолжительность жизни может достигать тридцати лет.
Болезнь Шильдера имеет такую симптоматику:
- снижение интеллекта;
- приступы эпилепсии;
- нарушение функционирования стриопаллидарной системы;
- тетрапарезы, возникающие как следствие гиперкинеза;
- признаки пигментного ретинита и гемералопии.
Диагностика

Для определения разновидности лейкодистрофии головного мозга потребуется комплексный подход, который основывается на инструментально-лабораторных исследованиях.
Однако не последнее место занимает первичная диагностика, которая включает в себя:
- изучение истории болезни как маленького пациента, так и его родителей – для выяснения пути наследования патологии;
- тщательный физикальный осмотр – для оценивания тонуса мышц, рефлексов, походки и координации движений. Сюда также стоит отнести консультации ЛОР-врача и офтальмолога – для определения наличия нарушений со стороны зрения или слуха;
- детальный опрос родителей пациента – для выяснения первого времени появления специфических признаков, поскольку в некоторых случаях очень важная информация касательно того, возникла ли симптоматика в младенческом возрасте или в ювенильном периоде.
Лабораторные исследования ограничиваются:
- анализом спинномозговой жидкости;
- биохимическими анализами крови – для выявления того, какие патологические вещества скапливаются при протекании того или иного варианта недуга.
Инструментальная диагностика уточняет тип болезни при помощи таких процедур:
- нейросонография;
- эхо-энцефалография;
- КТ и МРТ.
Также разработаны специфические методики ДНК-диагностики, обнаруживающие подобное заболевание ещё на этапе внутриутробного развития плода. В таких случаях необходима консультация специалиста по генетике.
Лечение
В настоящее время не существует эффективного лечения лейкодистрофии, позволяющего полностью устранить недуг. Пациентам показана симптоматическая терапия, которая в подавляющем большинстве случаев подразумевает проведение дегидратационной и антиконвульсивной терапии.
Единственной методикой лечения, помогающей продлить жизнь пациентам, является трансплантация пуповинной крови или пересадка донорского костного мозга. Однако это может занять от одного года до двух лет, на протяжении которых заболевание продолжает развиваться и прогрессировать. Именно по этой причине наступает либо инвалидизация пациента, либо летальный исход.
Необходимо отметить, что даже спешно проведённая трансплантация не изменит уже сформировавшиеся неврологические нарушения. Это лишь позволит замедлить процесс дальнейшего прогрессирования болезни.
На фоне того, что эффект от такого лечения наступает через большой промежуток времени, оно наиболее целесообразно только при раннем доклиническом диагностировании или при медленном прогрессировании такого расстройства.
Также стоит учесть, что трансплантация может привести к отторжению костного мозга, присоединению вторичных инфекций или к развитию синдрома «трансплантат против хозяина».
Профилактика и прогноз
Поскольку лейкодистрофия является генетически обусловленной болезнью, профилактических мероприятий, предупреждающих её развитие, не существует.
Единственной возможностью выявить недуг является пренатальная диагностика, т. е. проведённая в период вынашивания ребёнка – это поможет определить наличие лишь некоторых форм болезни, в частности, метахроматической лейкодистрофии.
Если в семье одного из родителей было зафиксировано такое заболевание, то перед тем, как завести ребёнка, паре необходимо пройти медико-генетическое консультирование.
Прогноз лейкодистрофии зачастую неблагоприятный – недуг приводит к глубочайшей деградации пациента и его смерти.
Лейкодистрофия в центральной нервной системе: причины, течение, прогноз
1. Что такое липидоз? 2. О наследственных лейкодистрофиях 3. Причины 4. Частота встречаемости 5. Течение 6. Диагностика 7. Клиническая картина 8. О терапии 9. Заключение
Кроме известных заболеваний центральной нервной системы, таких, как острые нарушения мозгового кровообращения, деменция, болезнь Паркинсона, существуют и редкие. К ним, например, относится такой патологический процесс в белом веществе, как лейкодистрофия. Он возникает по целом ряду причин и относится к наследственным липидозам. Что означают эти термины, как проявляются и лечатся эти заболевания?
Что такое липидоз?
Нервная система человека является высшим органом, координирующим вегетативные функции (питание, кровообращение, выделение, дыхание), сознательные мышечные движения, обеспечивающие взаимодействие человека с внешней средой. Венцом является высшая нервная деятельность и человеческое мышление, благодаря которому у вас появилась возможность читать этот текст на экране компьютера. Все это невозможно без нервного импульса, который создается нейронами, составляющими серое вещество коры больших полушарий головного мозга, подкорковых ядер и спинного мозга.

Именно потому, что липиды нерастворимы в воде, их мембраны полностью исключают потери импульсов, которые генерируются в водной среде цитоплазмы нейрона. Известно, что волна возбуждения, которая генерируется в нейроне с помощью работы натриево-калиевого насоса, может распространяться со скоростью более 100 метров в секунду.
Поэтому в нервной системе человека сосредоточено много миелина, который относится к липидам. Нарушение обмена и структуры липидов в головном мозге составляют заболевания, которые называют липидозами. Сюда входит группа наследственных лейкодистрофий веществаголовного мозга, о которых и пойдет речь.
Группа наследственных лейкодистрофий – это очень редкие заболевания, поэтому у врачей есть множество причин вначале подумать о более часто встречающейся патологии.
О наследственных лейкодистрофиях
В неврологии до середины 80-х годов XX века был принят термин «прогрессирующий склероз», в наше время он заменен более точным термином «лейкодистрофия». Лейкодистрофия – это группа заболеваний наследственного характера, которые характеризуются прогрессирующим поражением белого вещества, как головного, так и спинного мозга с разрастанием глиальных элементов и нарушением проведения нервного импульса.

Причины
Эти заболевания часто возникают при дефиците особых ферментов, которые участвуют в метаболизме липидсодержащих нервных структур. Так, метахроматическая лейкодистрофия своим существованием обязана дефициту арилсульфатазы, особого лизосомального фермента. При других вариантах возможно несовершенное образование миелина, или его распад. При лейкодистрофии Гринфилда (или поздней инфантильной форме) отсутствует фермент сульфатаза в моче.
Частота встречаемости
Практически все формы наследственных лейкодистрофий являются редкими заболеваниями, которые возникают не чаще, чем один случай на 40 тысяч человек, или даже реже. Наиболее часто возникает адренолейкодистрофия, следующая за ней метахроматическая лейкодистрофия встречается с частотой 1:55000, а глобоидный тип дистрофии Краббе встречается с частотой около 8-10 случаев на миллион. Есть и более редкие наследственные формы.
Течение
Все формы, возникающие в раннем детском возрасте, отличаются неуклонным прогрессированием, появлением новой симптоматики, и высокой возможностью летального исхода в раннем детстве. В том случае, если прогрессирование останавливается, то пациент может дожить и до 30 лет, правда, оставаясь глубоким инвалидом. Так, например, протекает лейкодистрофия Пелицеуса Мерцбахера, или ранняя инфантильная лейкодистрофия.
Часто, возникая в раннем детском возрасте, прогрессирование болезни приводит к смерти ребенка в возрасте от года до семи лет.
Известно, что чем позже начинаются симптомы болезни, тем легче течет болезнь. Например, метахроматическая лейкодистрофия типа Шольца, при выявлении ее у детей приводит к смерти через год – два после появления первых симптомов. Ювенильная форма, которая развивается в возрасте от 6 до 10 лет, приводит к летальному исходу через 4-6 лет. Поздняя лейкодистрофия, которая дебютирует в возрасте 18 лет, при относительно медленном прогрессировании, может позволить пациенту прожить до 30-40 лет, при очень благоприятном течении и отсутствии интеркуррентных заболеваний.

Тем не менее, большинство случаев приходится на ранний детский возраст, и, к сожалению с летальным исходом на 2-5 год жизни.
Диагностика
Обычно раздел диагностики принято помещать после клинической картины и перед лечением. Но липидозы, в связи с полиморфизмом симптомов и неуклонным прогрессированием до сих пор, несмотря на существование визуализирующих методов, таких, как КТ и МРТ, часто не поддаются прижизненной диагностике. Нужно помнить, что никакое исследование, даже МРТ с контрастированием в динамике, не в состоянии подтвердить этот процесс, а только «не исключить».
Это связано с тем, что симптомы лейкодистрофий чрезвычайно разнообразны, особенно у ребенка. Логика подсказывает, что скорее возможны с наибольшей степенью вероятности многие другие заболевания. У ребенка это могут быть проявления перинатального поражения центральной нервной системы, детского церебрального паралича, нейроинфекции. Как у детей, так и у взрослых, прежде всего врачам нужно исключать системные инфекционно – аллергические и аллергические поражения, опухоли.
Настоящие трудности возникают при попытке дифференцировать лейкодистрофии от демиелинизирующих заболеваний (оптикомиелита Девика, острого рассеянного энцефаломиелита, рассеянного склероза). На помощь могут прийти количественные анализы на определение дефектных липидов в крови и спинномозговой жидкости, а также малодоступные и очень дорогие генетические прицельные исследования, поскольку картина на МРТ не дает однозначного ответа.
Так, врач невролог, видя, например, фото кровоизлияния мозга, сразу поставит диагноз по характерным признакам. При наличии демиелинизирующих очагов нужно обращать внимание на клиническую картину, но, учитывая быстроту прогрессирования дистрофий белого вещества, и отсутствие разработанного лечения, зачастую оказывается, что окончательный диагноз выставляется только на вскрытии, особенно у маленьких детей.


Клиническая картина
Симптомы поражения очень многообразны. У ребенка можно встретить:
- диффузное снижение мышечного тонуса, с последующей сменой на гипертонус;
- появление дрожания головы, конечностей;
- судороги, немотивированное возбуждение, постоянный крик;
- глазодвигательные нарушения: косоглазие, нистагм, офтальмоплегия, как внутренняя, так и наружная;
- обратное развитие (дети утрачивают все приобретенные навыки);
На поздней стадии развиваются тяжелые параличи конечностей, бульбарные нарушения. Смерть наступает от паралича дыхательной мускулатуры, сосудодвигательного и дыхательного центра продолговатого мозга.
Метахроматическая лейкодистрофия может развиться в юношеском возрасте и у взрослых. В этом случае будут беспокоить:
- мозжечковые и экстрапирамидные нарушения (тремор, гиперкинезы, ригидность);
- атрофия зрительных нервов;
- центральные параличи и парезы;
- возникает выраженная деменция, возникают такие симптомы, как сенсомоторная афазия, или расстройство речи.
Нужно помнить, что метахроматическая лейкодистрофия иногда оставляет своим жертвам наибольшее время для жизни, но это время человек проживает тяжелым инвалидом, часто лишенный возможности не только передвигаться, но и мыслить.
О терапии
Специфического лечения даже такого длительно текущего заболевания, как метахроматическая лейкодистрофия во взрослой форме, не говоря уже о быстрых вариантах, поражающих структуры мозга ребенка, не существует. Существующее лечение сводится к введению гормонов, витаминов, поддержания функций мозга, пока человек может дышать.
Единственный шанс восстановить миелин и улучшить работу мозга в наше время – это аутотрансплантация стволовых клеток. Но даже при этом нужно длительный срок для его синтеза (по данным МРТ, год или два). Чаще всего, срок жизни при болезни значительно короче, особенно у детей.
Сравнить оголенные нервы, лишенные миелиновых оболочек, упакованные плотными пучками, можно лишь с энергосистемой целого города, провода и кабели которого не имеют изоляции и скручены между собой. В результате возникнет вспышка короткого замыкания с разрушением всей энергоструктуры. То же самое происходит при этих заболеваниях.
Лейкодистрофия как наследственное заболевание
Лейкодистрофия — это группа редких заболеваний, которые передаются наследственным путем. При этих патологических состояниях происходит разрушение миелиновых оболочек и распаду белого вещества в головном мозге. Передача болезни происходит по рецессивному и аутосомно-рецессивному типу, нарушенные хромосомы имеют сцепление с полом.
Поражения головного мозга и мозжечка при лейкодистрофиях происходит диффузно и симметрично. Серое вещество при этом практически не повреждается.
При данных заболеваниях выявляется дефект вещества, которое отвечает за липидный обмен, в частности, за синтез миелина. Обмен веществ нарушается повсеместно, поэтому его продукты могут обнаруживаться не только в ЦНС, но и других органах и тканях.
Типы заболевания
На данный момент существует несколько типов лейкодистрофий:
- Метахроматическая. Для нее характерной особенностью является интенсивная деструкция миелина и накопление токсичных продуктов извращенного обмена в ЦНС, нервных пучках и внутренних органах.
- Детская острая лейкодистрофия (болезнь Краббе). Она характеризуется преимущественным поражением миелина в спинном и головном мозге.
- Болезнь Галлевордена-Шпатца. Проявляется диффузным развитием склероза головного мозга.
- Лейкодистрофия Пелицеуса—Мерцбахера. При этой патологии существует четкая взаимосвязь передачи заболевания по половому признаку.
- Заболевание Канавана—ван Богарта—Бертранда. Для нее характерным является то, что процесс разрушения миелина начинается уже внутриутробно.
- Наиболее редким видом лейкодистрофии является болезнь Александера.
Как проявляются различные формы патологии
- При метахроматической лейкодистрофии первичная симптоматика проявляется в 2−3 года в виде двигательных расстройств и снижением тонуса мышц. Потом часто возникает судорожный синдром, а мышечный тонус повышается.
По мере прогрессирования заболевания развивается нарушение речи, снижение интеллектуальных способностей у ребенка, работы дыхательного и сосудистого центров. Обычно смерть наступает от присоединения инфекции в возрасте от 4 до 7 лет.
- Болезнь Краббе начинает манифестировать в возрасте от 4 месяцев, малыш становится чрезмерно возбудимым, и постоянно плачет. Приступы крика часто сопровождаются судорожным синдромом. Температура тела повышается при отсутствии признаков воспалительного процесса.
- Мышечный тонус повышается, развивается атрофия зрительных нервов. Происходит неуклонное прогрессирование заболевания, нарушается дыхательная функция и кровообращение. В терминальной стадии отмечается слабоумие, ригидность децеребрационного генеза и происходит полное истощение.
- Заболевание Галлевордена-Шпатца начинает проявлять первые признаки у детей от 7 до 12 лет в виде развития непроизвольных движений конечностей. Лейкодистрофия головного мозга приводит к тому, что постепенно растет ригидность мышц, атаксия, снижение интеллектуальных способностей.
Иногда бывают судороги. Болезнь склонна к медленному прогрессированию и может протекать на протяжении довольно длительного времени.
- Патология Пелицеуса—Мерцбахера начинается от 5 месяцев от рождения ребенка, но при этом прогрессирует достаточно медленно. Нарушается координация при движении, возникает неконтролируемое движение глазных яблок, дрожание головы. Постепенно начинается снижение зрения за чет атрофии нерва глаза, замедляется речь, ухудшаются интеллектуальные возможности.
Иногда встречается постепенное нарастание симптомов с последующим улучшением состояния в течение длительного времени. Описаны случаи заболевания без прогрессирования.
- Лейкодистрофия Канавана—ван Богарта—Бертранда проявляется уже сразу после появления на свет малыша. Он уже рождается вялый, сонливый, плохо есть и мало двигается. Иногда у него развиваются судороги. Когда ему исполняется 2−6 месяцев, тонус мышц шеи становится сниженным при повышении тонуса верхних и нижних конечностей.
Встречается непроизвольное движение глаз, гидроцефалия. Любое прикосновение приводит к состоянию опистотонуса. Быстрое ухудшение состояния приводит к полному расстройству жизненно важных функций и гибели в возрасте от полугода до двух лет.
- Лейкодистрофия Александера характеризуется увеличивающейся гидроцефалией, слабоумием, судорогами.
Как можно выявить наличие лейкодистрофии?
Поставить диагноз быстро и правильно при лейкодистрофии достаточно тяжело. Ее некоторые формы подтверждаются только после вскрытия.
Этапы диагностики при этой патологии должны быть такими:
- Обязательное и тщательное изучение анамнеза и симптоматики (первые признаки, быстрота их нарастания, скорость изменений).
- Поскольку лейкодистрофия является наследственным заболеванием, то тщательно анализируется семейный анамнез и наличие подобных случаев у близких родственников больного.
- При осмотре оценивается состояние мышц и их тонуса, наличие нормальных и патологических рефлексов, если малыш ходит, то изучаются особенности походки и координации при передвижении.
- По возможности проводится наблюдение за прогрессированием основных признаков в динамике. Изучаются нарушения зрения, глазные движения, слух, психическое развитие.
- Проводится пункция с забором ликвора, необходимо уточнить его цвет, давление, наличие белка и его количество, которое повышается в результате разрушения мозговых клеток, имеется ли цитоз. Оценивается наличие глюкозы и слей хлора.
- Обязательно используются биохимические методики исследования. Они позволяют оценивать уровня ферментных веществ, транспортировка которых нарушается при определенном виде лейкодистрофии. Или же находят токсические элементы, способные накапливаться при развитии данной патологии.
- Дополнительно при необходимости назначается прохождение КТ и МРТ с целью полного изучения головного мозга и степени его разрушения.
- Генетические тесты позволяют говорить о наследственной природе заболевания.
- В некоторых случаях проводятся современные методики диагностирования лейкодистрофии в пренатальном периоде.
При ведении больного иногда необходима консультация таких специалистов, как детский невролог и генетик.
Как помочь больному?
На данном этапе развития медицины не представляется возможным полное избавление от такого заболевания. Лечение проводится симптоматическое, направленное на улучшение качества жизни больного и снижение скорости прогрессирования процесса.