Что такое талассемия у мужчин. Талассемия, или почему нужно знать своих предков? Осложнения и последствия
Талассемия

Талассемия – наследственные гемоглобинопатии, характеризующиеся угнетением синтеза цепочечных белковых молекул, образующих структуру гемоглобина. Это приводит к повреждению мембраны эритроцитов и разрушению красных клеток крови с развитием гемолитических кризов. Признаками талассемии служат характерные костные изменения, гепатоспленомегалия, анемический синдром. Диагноз талассемии подтверждается клиническими и лабораторными данными (исследованием гемограммы, гемоглобина, миелограммы, электрофоретическим методом). Возможна пренатальная диагностика талассемии. В лечении талассемии применяются гемотрансфузии, терапия десфералом, спленэктомия, трансплантация костного мозга.
МКБ-10

Общие сведения
Талассемия – группа генетически детерминированных болезней крови, развивающихся при нарушении синтеза a- или β-цепей гемоглобина, сопровождающихся гемолизом, гипохромной анемией, микроцитозом. В гематологии талассемия относится к наследственным гемолитическим анемиям – количественным гемоглобинопатиям. Талассемия широко распространена среди населения Средиземноморского и Черноморского региона; название заболевания буквально переводится как «анемия морского побережья». Также случаи талассемии нередки в странах Африки, Ближнего Востока, Индии и Индонезии, Средней Азии и Закавказья. С синдромом талассемии каждый год в мире рождается 300 тыс. детей. В зависимости от формы патологии течение талассемии может быть тяжелым, фатальным или легким, бессимптомным. Так же, как серповидно-клеточная анемия, талассемия играет роль защитного фактора против малярии.
Классификация талассемии
С учетом поражения той или иной полипептидной цепи гемоглобина различают:
- a-талассемию (с подавлением синтеза альфа-цепей HbA). Данная форма может быть представлена гетерозиготным носительством манифестного (α-th1) или немого (α-th2) гена; гомозиготной a-талассемией (водянкой плода с гемоглобином Бартса); гемоглобинопатией Н
- b-талассемию (с подавлением синтеза бета-цепей HbA). Включает в себя гетерозиготную и гомозиготную β-талассемию (анемию Кули), гетерозиготную и гомозиготную δβ-талассемию (F-талассемию)
- γ-талассемию (с подавлением синтеза гамма-цепей гемоглобина)
- δ-талассемию (с подавлением синтеза дельта-цепей гемоглобина)
- талассемию, обусловленную нарушением структуры гемоглобина.
Статистически чаще встречается β-талассемия, которая, в свою очередь, может протекать в 3-х клинические формах: малой, большой и промежуточной. По тяжести синдрома выделяют легкую форму талассемии (пациенты доживают до половой зрелости), средне-тяжелую (продолжительность жизни больных 8-10 лет) и тяжелую (гибель детей наступает в первые 2-3 года жизни).
Причины талассемии
Талассемия является генетическим заболеванием с аутосомно-рецессивным наследованием. Непосредственной причиной патологии выступают различные мутационные нарушения в гене, кодирующем синтез той или иной цепи гемоглобина. Молекулярную основу дефекта могут составлять синтез аномальной матричной РНК, делеции структурных генов, мутации регуляторных генов либо их неэффективная транскрипция. Следствием подобных нарушений служит снижение или отсутствие синтеза одной из полипептидных гемоглобиновых цепей.
Так, при b-талассемии бета-цепи синтезируются в недостаточном количестве, что приводит к избытку альфа-цепей, и наоборот. Избыточно продуцируемые полипептидные цепи откладываются в клетках эритроидного ряда, вызывая их повреждение. Это сопровождается деструкцией эритрокариоцитов в костном мозге, гемолизом эритроцитов в периферической крови, гибелью ретикулоцитов в селезенке. Кроме этого, при b-талассемии в эритроцитах накапливается фетальный гемоглобин (НbF), не способный транспортировать кислород в ткани, что вызывает развитие тканевой гипоксии. Вследствие костномозговой гиперплазии развивается деформация костей скелета. Анемия, тканевая гипоксия и неэффективный эритропоэз в той или иной степени нарушают развитие и рост ребенка.
Для гомозиготной формы талассемии характерно наличие двух дефектных генов, унаследованных от обоих родителей. При гетерозиготном варианте талассемии пациент является носителем мутантного гена, унаследованного от одного из родителей.
Симптомы талассемии
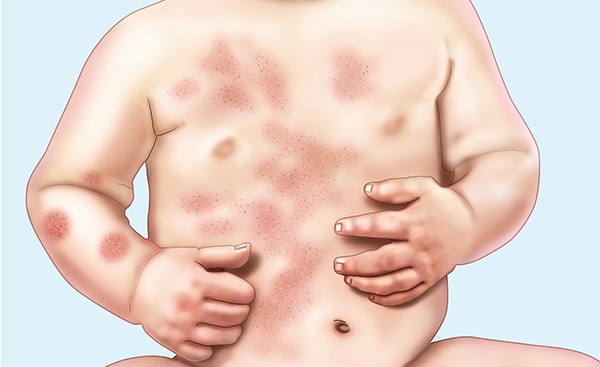
Признаки большой (гомозиготной) b-талассемии проявляются уже в течение 1-2-го года жизни ребенка. Больные дети имеют характерное монголоидное лицо, седловидную переносицу, башенный (четырехугольный) череп, гипертрофию верхней челюсти, нарушение прикуса, гепато- и спленомегалию. Проявлениями анемизации служат бледный или землисто-желтушный цвет кожных покровов. Поражение трубчатых костей сопровождается отставанием в росте и патологическими переломами. Возможно развитие синовита крупных суставов, калькулезного холецистита, язв нижних конечностей. Фактором, осложняющим течение b-талассемии, выступает гемосидероз внутренних органов, приводящий к развитию цирроза печени, фиброза поджелудочной железы и, как следствие, – сахарного диабета; кардиосклероза и сердечной недостаточности. Больные восприимчивы к инфекционным заболеваниям (кишечным инфекциям, ОРВИ и др.), возможно развитие тяжелых форм пневмонии и сепсиса.
Малая (гетерозиготная) b-талассемия может протекать бессимптомно или с минимальными клиническими проявлениями (умеренным увеличением селезенки, незначительно выраженной гипохромной анемией, жалобами на повышенную утомляемость). Аналогичная симптоматика сопровождает течение гетерозиготной формы a-талассемии.
При гомозиготной форме a-талассемии альфа-цепи полностью отсутствуют; фетальный гемоглобин у плода не синтезируется. Данная форма талассемии несовместима с жизнью, что приводит к внутриутробной гибели плода вследствие развивающегося синдрома водянки или самопроизвольному прерыванию беременности. Течение гемоглобинопатии Н характеризуется развитием гемолитической анемии, спленомегалии, тяжелых костных изменений.
Диагностика талассемии
Талассемию следует заподозрить у лиц с семейным анамнезом, характерными клиническими признаками и лабораторными показателями. Больные талассемией нуждаются в консультации гематолога и медицинского генетика.
Типичными гематологическими изменениями служат снижение уровня гемоглобина и цветового показателя, гипохромия, наличие мишеневидных эритроцитов, повышение уровня железа сыворотки крови и непрямого билирубина. Электрофорез Hb на ацетат-целлюлозной пленке используется для определения различных гемоглобиновых фракций. При изучении пунктата костного мозга обращает внимание гиперплазия красного кроветворного ростка с высоким числом эритробластов и нормобластов. Молекулярно-генетические исследования позволяют выявить мутацию в локусе a- или β-глобина, нарушающую синтез полипептидной цепи.
На краниограммах при большой b-талассемии выявляется игольчатый периостоз (феномен «волосатого черепа»). Характерна поперечная исчерченность трубчатых и плоских костей, наличие мелких очагов остеопороза. С помощью УЗИ брюшной полости обнаруживается гепатоспленомегалия, камни желчного пузыря.
При подозрении на талассемию требуется исключить железодефицитную анемию, наследственный микросфероцитоз, серповидно-клеточную анемию, аутоиммунную гемолитическую анемию. В семьях, имеющих больных талассемией, рекомендуется проведение генетического консультирования супругов и инвазивной дородовой диагностики (биопсии хориона, кордоцентеза, амниоцентеза) для выявления гемоглобинопатии на ранних сроках беременности. Подтверждение гомозиготных форм талассемии у плода служит показанием для искусственного прерывания беременности.
Лечение и прогноз талассемии
Лечебная тактика при различных формах талассемии неодинакова. Так, пациенты с малой b-талассемией в лечении не нуждаются. С другой стороны, больным с гомозиготной b-талассемией с первых месяцев жизни требуется проведение гемотрансфузионной терапии (переливание размороженных или отмытых эритроцитов), введение хелатирующих препаратов, связывающих железо (дефероксамина), глюкокортикоидов при возникновении гемолитических кризов. При всех формах талассемии показан прием препаратов фолиевой кислоты и витаминов группы В.
При гиперспленизме (особенно на фоне гемоглобиноза Н) требуется удаление селезенки (спленэктомия). Из-за склонности к присоединению инфекционных осложнений больным рекомендуется обязательная вакцинация против пневмококковой инфекции. Многообещающим методом лечения талассемии служит трансплантация костного мозга от гистосовместимого донора.
Прогноз больших форм талассемии неблагоприятный; больные погибают в младенческом или молодом возрасте. При гетерозиготной бессимптомной форме талассемии продолжительность и качество жизни в большинстве случаев не страдают. Первичная профилактика талассемии включает предупреждение браков между гетерозиготными носителями генов заболевания, а при высоком генетическом риске рождения больного потомства – отказ от деторождения.
Талассемия

Талассемия – это название группы наследственных заболеваний крови. При талассемии организм вырабатывает меньше эритроцитов и гемоглобина, чем в нормальном состоянии.
Что такое талассемия?
Талассемия – это название группы наследственных заболеваний крови. При талассемии организм вырабатывает меньше эритроцитов и гемоглобина, чем в нормальном состоянии. Гемоглобин – это богатый железом белок, содержащийся в эритроцитах (красных кровяных тельцах), позволяющий им переносить кислород из легких по всему организму. Недостаток гемоглобина в эритроцитах называют анемией. Анемия нарушает способность организма доставлять кислород из легких в конечности и другие части тела.
Каковы симптомы талассемии?
Симптомы талассемии зависят от типа заболевания и тяжести анемии. У одних людей симптомов либо нет вообще, либо они незначительны. У других же симптомы носят умеренный или тяжелый характер. В целом, к наиболее распространенным признакам заболевания относятся:
- Бледность
- Усталость, отсутствие сил или мышечная слабость (утомляемость)
- Головокружение или затрудненное дыхание
- Отсутствие аппетита
- Темный цвет мочи
- Желчность (пожелтение кожи и белков глаз)
- У детей – медленное развитие и отложенный период полового созревания
- Костные деформации на лице
- Вздутие живота
У детей с врожденной талассемией ее симптомы могут проявиться как сразу же, так и через какое-то время. Как правило, большинство симптомов проявляется в течение первых двух лет жизни. Если вы заметили, что ребенок развивается медленнее сверстников, очень важно выяснить возможность наличия у него талассемии, поскольку при отсутствии должного лечения это заболевание способно привести к сердечной недостаточности и инфицированию.
Каковы причины развития талассемии?
Талассемия передается путем наследования генной мутации от одного или обоих родителей. Эти мутировавшие гены обусловливают более высокий темп потери эритроцитов и снижают выработку гемоглобина.
Конкретный тип талассемии зависит от того, какие именно мутировавшие гены человек наследует от родителей и каково их общее количество. Существует 2 основных типа талассемии: альфа-талассемия и бета-талассемия, названные так, соответственно, из-за альфа-глобина и бета-глобина – белков, отвечающих за выработку гемоглобина.
- Альфа-талассемия: Этот тип заболевания задействует 4 гена (2 от матери и 2 от отца). Если вы унаследуете лишь 1 ген, то лично у вас не проявится никаких признаков талассемии, но впоследствии вы сохраните способность передать генную мутацию своим детям. При этом вы сами являетесь носителем. При наличии двух мутировавших генов симптомы будут умеренными, трех – от умеренных до тяжелых, а вот ребенок, унаследовавший все 4 гена, родится очень больным и, скорее всего, умрет вскоре после рождения.
- Бета-талассемия: В этом типе заболевания участвуют 2 гена (1 от матери и 1 от отца). При наследовании только 1 гена симптомы талассемии будут умеренными, а при наследовании сразу 2 – от умеренных до тяжелых, причем проявятся они, скорее всего, в первые 2 года жизни.
Кто входит в группу риска?
В группу риска входят люди, у которых в роду были случаи талассемии. Этому заболеванию в равной степени подвержены как мужчины, так и женщины, но у представителей определенных этнических групп наблюдается повышенная предрасположенность к талассемии:
- Альфа-талассемия наиболее часто встречается среди выходцев из Юго-Восточной Азии, Индии, Китая или Филиппин.
- Бета-талассемия чаще наблюдается у выходцев из Средиземноморья (Греции, Италии и Восточной Европы), Азии и Африки.
Как диагностируется талассемия?
Если врач заподозрил у вас или вашего ребенка талассемию, он проведет осмотр и задаст вопросы о вашей наследственности. Талассемия диагностируется исключительно по результатам анализа крови, причем для определения наличия заболевания используются сразу несколько видов этого анализа. Одни измеряют количество и размер эритроцитов или содержание железа в крови. Другие оценивают количество гемоглобина в эритроцитах. Тест ДНК помогает врачу точно определить поврежденные или отсутствующие гены.
Лечение
Лечение талассемии зависит от типа заболевания и тяжести симптомов. Если у вас отсутствуют симптомы или их тяжесть умеренная, вам может понадобиться либо минимальное лечение, либо отсутствие такового.
Лечение умеренной и тяжелых форм талассемии зачастую включает регулярные переливания крови и прием фолиевых добавок, помогающих организму вырабатывать здоровые кровяные тельца. Альфа-талассемию иногда ошибочно принимают за железодефицитную анемию, и в таком случае в качестве лечения назначаются пищевые добавки, обогащенные железом. Однако они оказываются абсолютно неэффективными против талассемии.
При частых переливаниях в крови может накопиться слишком большое количество железа. В таком случае проводится так называемая хелатирующая терапия, позволяющая вывести лишнее железо из организма. Поэтому, если вы подвергаетесь переливаниям, не следует принимать железосодержащие добавки.
В наиболее тяжелых случаях может помочь трансплантация костного мозга или стволовых клеток для замены поврежденных клеток на здоровые донорские, которые обычно берутся у ближайшего родственника (брата или сестры).
Как жить с талассемией?
Хотя вы никак не можете предотвратить наследование талассемии, вы способны при помощи определенных действий максимально приблизить качество жизни к оптимальному. К таковым относятся:
- Четкое следование плану лечения. Делайте переливания крови настолько часто, насколько рекомендует врач, принимайте фолиевую кислоту и/или проходите хелатирующую терапию.
- Беспрерывное лечение. Регулярно посещайте врача для прохождения осмотров и сдавайте все назначенные им анализы. Это могут быть как специализированные тесты, относящиеся к талассемии, так и индикаторы общего состояния здоровья. Не забывайте прививаться от гриппа, пневмонии, гепатита В и менингита.
- Здоровый образ жизни. Питайтесь согласно составленному плану. Во время пика сезонных заболеваний регулярно мойте руки и избегайте скоплений людей во избежание инфицирования. Следите за чистотой в помещении, особенно в местах проведения переливания. При появлении жара или других признаков инфекции сразу же обратитесь к врачу.
- Группы поддержки. Присоединитесь к существующим в вашем регионе группам поддержки, чтобы делиться с другими людьми опытом жизни с талассемией. Но ни в коем случае не вносите коррективы в план лечения без предварительного одобрения со стороны лечащего врача.
Какие могут быть осложнения?
Талассемия может привести к развитию других проблем со здоровьем:
- Увеличениеселезенки. Селезенка помогает организму бороться с инфекциями и фильтровать поврежденные клетки крови. При талассемии работа селезенки может стать более интенсивной, в результате чего она увеличивается в размерах. При критическом увеличении селезенка зачастую подлежит удалению.
- Инфекции. Люди с талассемией более склонны к заражению инфекциями крови, особенно при частых переливаниях. Некоторые типы инфекций усугубляются в виду удаления селезенки.
- Костные проблемы. Талассемия может привести к костным деформациям лица и черепа. Также у страдающих этим заболеванием людей часто развивается тяжелый остеопороз (ломкость костей).
- Переизбытокжелезавкрови. Это нарушение может в свою очередь нанести ущерб сердцу, печени и эндокринной системе (например, щитовидной и надпочечниковой железам).
Что делать, если я являюсь носителем талассемии и хочу при этом забеременеть?
Некоторые особенно тяжелые типы талассемии приводят к смерти ребенка еще до рождения или вскоре после него. Если вы или ваш партнер знаете, что являетесь носителями талассемии, вам нужно обязательно проконсультироваться с профильным специалистом или генным консультантом перед зачатием. Определенные тесты могут показать, переносчиком какого именно типа заболевания вы являетесь. Забеременев, вы сможете узнать о наличии или отсутствии у плода талассемии при помощи предродового тестирования.
Всё о талассемии
Система кровообращения – одна из самых важных в человеческом организме. Её нормальная работа зависит от множества факторов, важнейшим из которых является гемоглобин, уровень содержания которого в крови считается важнейшим клиническим показателем. Если же по тем или иным причинам синтез этого сложного животного белка нарушается, падает насыщаемость тканей и органов человека кровью, что в свою очередь может спровоцировать в них развитие патологических изменений. Талассемия, о которой мы сегодня будем говорить, — одна из таких патологий.
Разбираемся в теории
Талассемия, которую иногда называют анемией Кули, не принадлежит к числу распространённых заболеваний. Это редко встречающаяся наследственная патология, при которой нарушается синтез полипептидных цепей, являющихся основой нормального гемоглобина. Проще говоря, молекула этого сложного железосодержащего белка состоит из двух «частей»: красящего вещества и комплекса из 2 альфа-цепей и 2 бета-цепей. Следовательно, даже незначительная проблема, которая может негативно повлиять на синтез одной из составляющих гемоглобина, нарушит работу всей кровеносной системы с самыми негативными последствиями для организма в целом. Именно такой сбой и называют талассемией.

Типы заболевания
Приведённая выше схема несколько упрощённая, потому разговор о классификации талассемии придётся начать с небольшого дополнения, касающегося строения гемоглобина. Дело в том, что оно описывает так называемый тип A (Hb A, или A1), формула которого A2B2 (по две альфа- и бета цепи): его в крови около 90%. Ещё примерно 2-2,5% приходиться на гемоглобин A2 (Hb A2, A2D2). В нём вместо бета- присутствуют дельта-цепи. Оставшиеся 7-7,5% — это тип Hb A3, или Hb F (A2G2, его ещё называют плодный или фетальный), где место бета-цепей заняли гамма-цепи. Следовательно, классификация форм талассемии, если в её основу положить локализацию поражённого участка, должна выглядеть следующим образом.
Альфа-талассемия, самая распространённая:
- По гетерозиготному носительству манифестного гена альфа-th1
- По гетерозиготному немому гену альфа-th
- Альфа-талассемия (гомозиготная, её часто называют водянкой плода).
- Гемоглобинопатия H.
Также возможны бета-, гамма-, дельта- и смешанный бета-дельта варианты, которые структурно никак не подразделяются. Иногда встречается талассемия, обусловленная глобальными (структурными) нарушениями гемоглобина:
- Leporo-гемоглобинопатия.
- Constant Spring (GS) гемоглобинопатия.
Классификация на основе фактора наследования:
- от обоих родителей (гомозиготная форма).
- от одного родителя (гетерозиготная форма).
Классификация по клинической форме:
- Большая бета-талассемия. Наиболее опасные осложнения – нарушения местного кровообращения из-за дефекта кожи, глубокие нарушения функций печени (цирроз), фиброз поджелудочной железы, часто сопровождаемая симптомами сахарного диабета.
- Промежуточная бета-талассемия. Обычно протекает не так тяжело, а клинические проявления менее выражены. Основные осложнения – гипертрофия селезёнки и поражение костной системы.
- Малая бета-талассемия. Снижение уровня красных кровяных телец и гемоглобина незначительно, а проблемы со здоровьем ограничены лёгкой анемией.
- Минимальная бета-талассемия, или Синдром Сильверстони-Бьянко. Распознать её можно по результатам специального генетического исследования. Чаще всего протекает совершенно бессимптомно и никаких неприятностей человеку не доставляет.
Классификация по степени тяжести клинических проявлений:
- Тяжёлая. Чаще всего она обнаруживается у новорождённых, причём вероятность летального исхода крайне высока.
- Хроническая со средней тяжестью. Пациенты обычно доживают до 7-10 лет, но дальнейшие прогнозы малоутешительны.
- Хроническая с лёгкой тяжестью. Большинство больных ведут практически нормальную жизнь, а критические проблемы со здоровьем начинаются уже в зрелом возрасте.
Возможные причины
Как мы уже выяснили, талассемия обусловлена наследованием мутантного гена по аутосомно-рецессивному типу. Потому заболеть могут дети, у которых предрасположенность к талассемии имеют оба родителя. Другими словами, если они получили соответствующий ген и от матери, и от отца. В том случае, если аномалия наблюдается у одного из них, вероятность заболеть снижается (от 25% до 50%), а клинические проявления будут либо слабовыраженными, либо отсутствовать полностью.

Но основная проблема талассемии (как и многих других наследственных патологий) в том, что здоровый геном может мутировать. Или, говоря по другому, вероятность «заболеть» в данном случае все же имеется, хотя она и достаточно мала. Какие же факторы могут спровоцировать мутацию?
- Ионизирующее излучение и повышенный радиационный фон. Для этого совсем необязательно жить возле атомного реактора: достаточно, к примеру, слишком активно пользоваться мобильным телефоном или по много часов сидеть возле монитора компьютера. Да и Чернобыль, хотя авария случилась почти 30 лет назад, ещё долго будет напоминать о себе. Такой эффект может ещё дать усиленная радиотерапия при онкологических заболеваниях или многократные рентгенографические исследования (особенно, на устаревшей технике).

- Различные химические вещества с сильным мутагенным эффектом. В группе риска работающие на вредных производствах, хотя сбрасывать со счетов чрезмерное увлечение недоброкачественными продуктами питания тоже не стоит.
- Фармакологические препараты. Спровоцировать мутацию могут и сильнодействующие лекарства. В основном это цитостатики (препараты, направленные на борьбу со злокачественными новообразованиями) прошлых поколений.
- Вирусы: герпеса, кори или даже обычного гриппа.
- Курение. Главные виновники – вредные химические соединения, содержащиеся в табачном дыме (фенол, бензол).
- Низкокачественный дешёвый алкоголь или злоупотребление крепкими напитками.
Симптомы и клинические проявления
- Седловидная (сильно сплющенная) переносица.
- Патологически увеличенная верхняя челюсть.

- Выраженная бледность или желтушность кожных покровов.
- Форма черепа близка к квадратной.
- Сильное суженый разрез глаз даже у представителей европеоидной расы.
- Увеличенная селезёнка и печень.
- Билирубиновые камни в жёлчных путях.
- Значительное отставание в развитии (как физическом, так и половом).
- Повышенная утомляемость и слабость даже при отсутствии физических нагрузок.
- Язвочки в зоне голеней.
- Водянка плода.
- Падение иммунного ответа, из-за чего организм перестаёт сопротивляться вирусам и бактериям.

Методы дифференциальной диагностики
- Углублённый осмотр.
- Общий анализ крови: уровень гемоглобина ниже 50 г/л, гипохромные эритроциты, цветовой показатель не более 0,5, много ретикулоцитов (2,5-4%) и железа.
- Биохимия крови: гипербулирубинемия, гиперсидеремия, ОЖСС (общая железо-связывающая способность сыворотки).
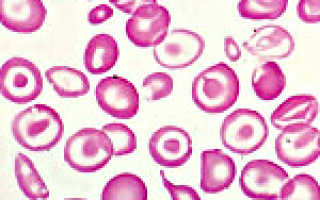
- Мазок крови. Анизоцитоз, пойкилоцитоз, помимо гипохромных, видны малые и мишеневидные эритроциты.
- Электрофорез на ацетат-целлюлозной плёнке даёт увеличенный уровень фетального гемоглобина.
- Исследование цепей биосинтеза глобина в пробирке.
- Пункция костного мозга: увеличение числа сидеробластов.
- Рентгенография поможет обнаружить остеопороз костей черепа (синдром «щётки и ёжика») и поперечную исчерченность мелких костей кистей и стоп.
- ПЦР-диагностика выявит мутацию в локусе бета-глобина в 11 паре хромосом.

Внимание! При альфа-талассемии симптомы чаще всего менее отчётливы, чем при бета-форме.
Лечение
Несмотря на бурное развитие медицины, говорить о полном излечении талассемии не приходится, потому все проводимые мероприятия направлены на улучшение качества жизни больного и максимальное продление его жизни.
- При тяжёлых формах талассемии показано переливание цельной крови или эритроцитовой массы. Это даёт временный эффект, а риск гемосидероза при этом значительно увеличивается.
Отбор эритроцитарной массы ведётся с помощью технологии эртироцитафереза: https://krasnayakrov.ru/donorstvo/eritrocitoferez.html
- Переливание отфильтрованных, отмытых или размороженных эритроцитов вызывает гораздо меньше побочных эффектов, особенно если оно дополняется введением хелатов железа. Последняя методика может значительно увеличить срок жизни больного, но для поддержания нормального (относительно) состояния к ней придётся прибегать 5 раз в неделю на протяжении многих лет.

- Введение глюкокортикоидов при гемолитических кризах.
- Спленоэктомия (удаление селезёнки). Оптимальный возраст для операции – 8-10 лет. К сожалению, заметный эффект длится не более 1-2 лет, после чего наступает ухудшение, а риск проникновения в организм инфекций увеличивается.
- Длительный (не менее месяца) приём гепатопротекторов для поддержания функций печени.
- Трансплантация (пересадка) костного мозга. Единственный известный сегодня метод, который обеспечивает радикальное излечение от талассемии. Но из-за высокой стоимости операции и весьма длительного поиска подходящего донора рассчитывать на него не приходится, причём многие пациенты ждут своей очереди по несколько лет.
- Витамин C (аскорбиновая кислота) ускоряет выведение железа из организма.
- Специальное диетическое питание на протяжении всей жизни. Главный принцип – максимальное насыщение рациона танином, который значительно уменьшает всасывание железа (подойдут какао, чай, соя и орехи).
Талассемия у детей и беременных
Это заболевание, как мы уже выяснили, практически всегда диагностируется либо в роддоме, либо в первые годы жизни. Потому талассемию с некоторой натяжкой можно назвать детской, а все известные методы лечения относятся именно к этой возрастной категории. Случаев, когда этот диагноз ставился взрослым, единичны и никак не влияют на общую картину. Немногочисленные отличия, касающиеся терапии детской талассемии, касаются лекарственных препаратов. Их дозировка обычно несколько меньше, а спектр разрешённых к применению – немного уже. Но, повторимся, значимых и существенных отличий нет.
С беременными ситуация аналогичная. Если женщина с талассемией решается завести ребёнка, ей необходимо регулярно посещать гинеколога:
- I триместр: 1 раз в месяц.
- II триместр: 1 раз в 2-3 недели.
- III триместр: каждые 7-10 дней.
Если состояние стабильное, от использования сильнодействующих медикаментов лучше воздержаться, а к рискованным методам лечения можно прибегать в том случае, если иного выхода нет, и стоит вопрос о сохранении жизни. На учёт в женскую консультацию следует встать как можно раньше.
Прогноз
Качество жизни пациента, как и продолжительность его жизни, зависит от многих факторов. При этом – важное замечание – с некоторыми из них медицина научилась успешно бороться, а другие чаще всего удаётся купировать. Малая и минимальная формы (бета-талассемия) – это чаще всего обычная продолжительность жизни. При большой и промежуточной талассемии прогноз значительно хуже, а пациенты не всегда доживают до совершеннолетия. Но здесь следует понимать, что всё это верно для тех случаев, когда по тем или иным не проводилась трансплантация костного мозга. Если же удаётся найти донора, а родственники (чаще всего – родители) соберут необходимую сумму, то удаётся добиться либо полного излечения, либо значительного продления жизни.
Профилактические мероприятия
- Обязательная пренатальная диагностика.

- Если один из родителей – носитель мутантного гена, с помощью фетоскопии и амниоцентеза получают генетический материал плода, который затем подвергается генетическому исследованию, и в случае подтверждения предварительного диагноза рассматривается вопрос о прерывании беременности. Помните, что данные методы не избавлены от серьёзных побочных эффектов и требуют высокой квалификации медицинского персонала.
Талассемия – заболевание очень опасное и часто заканчивается летальным исходом. Но при своевременной диагностике, взвешенном подходе к планированию беременности и квалифицированной медицинской помощи вероятность смертельного исхода можно снизить в несколько раз. И хотя полное излечение, если не принимать в расчёт пересадку костного мозга или биомедицинские технологии на основе стволовых клеток, пока ещё невозможно, предаваться унынию и ставить крест на таком пациенте нельзя.
Талассемия
. или: Анемия Кули
Талассемия – это группа наследственных заболеваний, при которых происходит нарушение синтеза какой-либо цепи гемоглобина. Гемоглобин состоит из 2-х частей: гема и глобина. Гем (красящее вещество) представляет собой небелковую часть гемоглобина, в своем составе содержит железо. Глобин – белковая часть гемоглобина, которая представляет собой комплекс из 4-х полипептидных (белковых) цепей: 2 альфа-цепи и 2 бета-цепи.
Симптомы талассемии
Формы
Причины
Врач терапевт поможет при лечении заболевания
Диагностика
- Общий осмотр: башенный череп, седловидная переносица, монголоидный разрез глаз, увеличение печени и селезенки, желтушность и бледность кожных покровов, язвы в области голеней, отставание в физическом и половом развитии.
- Анализ крови: снижение гемоглобина до 30-50 г/л, гипохромные эритроциты (красные клетки крови окрашены слабо вследствие низкого содержания в них гемоглобина), цветовой показатель (степень насыщения эритроцитов гемоглобином) 0,5 и ниже, увеличение ретикулоцитов (предшественники эритроцитов) до 2,5-4%, повышение железа сыворотки (жидкая часть) крови.
- Мазок крови: гипохромные эритроциты (слабо окрашенные) малых размеров (диаметр менее 7-8 мкм), мишеневидные (клетки с бледной тонкой периферией и центральным утолщением); характерен анизоцитоз (клетки разного размера) и пойкилоцитоз (изменение формы эритроцитов — от правильной, округлой, до овальной, серповидной и т.п.).
- Биохимия крови: гипербилирубинемия (повышение уровня билирубина за счет свободной фракции), гиперсидеремия (перегрузка железом), снижение общей железосвязывающей способности сыворотки (ОЖСС).
- Электрофорез гемоглобина на ацетат-целлюлозной пленке (pH 9,0 и 6,5) с последующим количественным определением гемоглобиновых фракций. При гомозиготной талассемии уровень фетального гемоглобина (гемоглобин плода; у взрослого человека в крови содержится лишь 1%) увеличен.
- Изучение биосинтеза цепей глобина in vitro (в пробирке).
- Пункция костного мозга: повышенное содержание сидеробластов (незрелые формы эритроцитов, которые имеют ядра).
- Рентгенологическое исследование костей: для большой β-талассемии характерны мелкие участки остеопороза (снижение плотности костной ткани) наряду с участками гипертрофии (увеличение объема и массы) костей черепа — так называемый симптом щетки или ежика, а также поперечная исчерченность мелких костей стоп и кистей.
- Молекулярное исследование (ПЦР), с помощью которого определяют мутацию в локусе β -глобина на 11-й паре хромосом, нарушающую синтез β -глобиновой цепи.
- Клинические и лабораторные данные при альфа-талассемии выражены менее отчетливо, чем при β-талассемии.
- Возможна также консультация гематолога, медицинского генетика.
Лечение талассемии
- При тяжелых формах (например, при большой β-талассемии) переливание цельной крови (кровь целиком, то есть жидкая часть в сочетании с белками и клетками крови) или эритроцитарной массы (отдельно эритроцитов) дают лишь временный эффект, кроме того, при них возрастает опасность гемосидероза (отложение пигмента, содержащего оксид железа).
- В настоящее время наиболее эффективным считается переливание размороженных, отмытых или фильтрованных эритроцитов, которые гораздо реже вызывают побочные реакции, с одновременным длительным введением хелатов железа.
- Хелат железа должен вводиться длительно (в течение нескольких часов) подкожно. Поэтому созданы специальные аппараты (помпы), которые фиксируются к одежде. Больной с тяжелой формой заболевания должен получать препарат 5 дней в неделю на протяжении всей жизни. Для того чтобы не произошло местного повреждения тканей, необходимо периодически менять места инъекций. При возникновении гемолитических кризов необходимо вводить глюкокортикоиды в небольших дозах.
- При больших размерах селезенки проводят ее удаление (спленоэктомия). Операцию не следует делать детям до 5 лет. Оптимальный возраст — 8-10 лет. Хороший эффект обычно наблюдается в течение первого года после удаления, затем снова возникает ухудшение. Также возрастает риск инфекционных заболеваний.
- В настоящее время наиболее предпочтительным считается пересадка (трансплантация) костного мозга. Это единственный метод радикального лечения талассемии. Однако найти подходящего донора обычно сложно.
- Больным следует соблюдать диету, употреблять продукты, которые содержат танин: чай, какао, а также орехи, сою. Эти продукты уменьшают всасывание железа.
Также необходимо симптоматическое лечение:
- для улучшения функции печени необходимо назначение гепатопротекторов продолжительностью не менее месяца;
- назначение аскорбиновой кислоты (витамин С) способствует выведению железа из организма.
Осложнения и последствия
Профилактика талассемии
- Первичная профилактика включает в себя пренатальную (дородовую) диагностику.
- Если отец и мать страдают талассемией, целесообразно исследование плода во время беременности на предмет заболевания талассемией с целью возможного своевременного прерывания беременности. Применяют два метода: фетоскопию и амниоцентез. С помощью них получают клетки плода (делают пункцию (прокол) через переднюю брюшную стенку, первый метод проводят под контролем УЗИ) и после проводят их медико-генетическое исследование. Данные методы считаются опасными, так как могут вызвать преждевременные роды, инфицирование и даже гибель плода.
- Родителям, у которых есть родственники с данной патологией, необходимо обратится до планирования беременности к генетику, чтобы тот назначил необходимое дородовое обследование.
Дополнительно
Что делать при талассемии?
- Выбрать подходящего врача терапевт
- Сдать анализы
- Получить от врача схему лечения
- Выполнить все рекомендации