Врождённая сидеробластная анемия с экзокринной недостаточностью поджелудочной железы (Синдром Пирсона). Синдром Кларка-Хэдвилда. Клинико-психологический подход к проблеме митохондриальной патологии Синдром пирсона диагностика семьи
Врождённая сидеробластная анемия с экзокринной недостаточностью поджелудочной железы (Синдром Пирсона). Синдром Кларка-Хэдвилда
Синдром Пирсона
Врождённая сидеробластная анемия с экзокринной недостаточностью ПЖ была впервые описана Н.А. Pearson в 1979 г. Учёные более десятилетия наблюдали 4 детей, у которых отмечали рефрактерную сидеробластную анемию, вакуолизацию клеток предшественников костного мозга, экзокрипную панкреатическую недостаточность. Явные отличия по картине крови и изменениям в костном мозге от синдрома Швахмана позволили авторам выделить этот синдром в отдельную форму, которую по мере появления всё более новых случаев синдрома стали называть синдромом Пирсона.
Лишь десятилетием позже удалось определить, что в основе синдрома лежит генетический дефект, который проявляется множественными делениями и дупликациями митохондриальной ДНК, при этом следует отметить, что семейный анамнез у больных с синдромом Пирсона, как правило, не отягощен, что предполагает спорадичность мутации.
Существуют лишь единичные случаи, когда одну и ту же делению выявляли у ребёнка, страдающего синдромом Пирсона и у матери, страдающей офтальмоплегией, что возможно свидетельствует о более широком фенотипическом проявлении синдрома Пирсона, чем это принято до сих пор считать.
Изменение структуры митохондриальной ДНК при синдроме Пирсона отмечают не только в костном мозге и ациноцитах, но и в других органах — почках, сердце, печени, скелетных мышцах. При этом согласно ряду описаний, частота регистрируемых изменений структуры митохондриальной ДНК колеблется в пределах 50—95%. Гистологически поражение печени характеризуется накоплением железа в гепатопитах; поражение почек — вакуолизацией эпителия почечных канальцев, склерозом клубочков, проксимальной тубулопатией, развитием множественных кортикальных кист. Развитие фиброза в отдельных случаях наблюдают и в сердечной мышце, что клинически проявляется сердечной недостаточностью, как правило, левожелудочковой.
В ряде случаев прижизненная диагностика изменений митохондриальной ДНК может не давать положительных результатов. В то же время картина периферической крови весьма специфична: отмечают тяжёлую макроцитарную анемию с пейтропенией и тромбопитопенией. Эффекта от применения цианокобаламина, препаратов железа не наблюдают. С целью коррекции анемии большинство больных получают заместительную трансфузионную терапию.
Картина костного мозга представлена уменьшенным цитозом, вакуолизированными эритро- и миелобластами, кольцевидными сидеробластами. Имеются единичные описания, в которых типичная гематологическая картина появлялась только на 2-м году жизни, в то время как ранее доминировал синдром экзокринной панкреатической недостаточности.
Для данного синдрома характерно прогрессирующее увеличение делеций митохондриальной ДНК в течение жизни, при этом отмечена определённая динамика увеличения со временем частоты возникновения делеций, что, возможно, и определяет прогрессирующее течение болезни и спектр клинических проявлений заболевания.
В основе экзокринной недостаточности ПЖ при этом синдроме лежит врождённый дефицит секреции панкреатических амилазы, липазы и бикарбонатов. Морфологически структура ПЖ представлена атрофированной ацинарной тканью и фиброзными изменениями. При выраженных структурных изменениях ПЖ отмечают инсулино-потребный сахарный диабет, в том числе и в неонатальном периоде. Следует отметить, что есть единичные сообщения, свидетельствующие о возможности отсутствия клинически выраженной панкреатической недостаточности в первые месяцы и годы жизни, однако в большинстве случаев отмечают стеаторею, диарею и лактоацидоз.
Прогноз заболевания неблагоприятный, у большинства пациентов наблюдают замедление физического, в том числе внутриутробного развития, и больные погибают в течение первых месяцев или лет жизни, редко доживая до подросткового возраста. Характер и тяжесть течения заболевания могут определяться числом и локализацией делений ДНК, а также темпами их появления с возрастом.
Синдром Кларка-Хэдвилда
Синдром Андерсена
Синдром Андерсена связан с генетически детерминированным дефицитом амилотрансглюкозидазы. Синдром клинически напоминает муковисцидоз, но протекает значительно тяжелее, как правило, заканчиваясь летальным исходом в первые 5—6 лет жизни. Клинически характеризуется гепатомегалией, прогрессирующей атрофией ПЖ, стеатореей, гиповитаминозами, анемией, бронхоэктазами, эозинофилией, глюкозурией, отставанием в физическом развитии и отёчно-асцитическим синдромом.
Наследственная сидеробластная анемия
Рубрика МКБ-10: D64.0
Содержание
Определение и общие сведения [ править ]
Сидеробластные анемии представляют собой группу редких гетерогенных наследственных или приобретенных нарушений костного мозга, которые могут быть изолированными или быть частью некоторых синдромов. Сидеробластные анемии характеризуются снижением синтеза гемоглобина из-за нарушения утилизации железа (уровни плазмтического железа могут быть нормальными или повышенными), наличием ринг-форм (кольцевидных) сидеробластов в костном мозге из-за патологической перегрузки железом митохондрий, которые визуализируются окрашиванием Перлса.
Группа включает приобретенные (идиопатические) сидеробластные анемии и конституциональные сидеробластные анемии. Последние включают в себя синдромальные сидеробластные анемии, такие как синдром Пирсона, митохондриальную миопатию с сидеробластной анемией, Х-сцепленную сидеробластную анемию с атаксией, тиаминовый мегалобластный анемический синдром и несиндромальные сидеробластные анемии: Х-сцепленную и аутосомно-рецессивную сидеробластные анемии.
Cиндром Пирсона характеризуется наличием рефрактерной сидеробластной анемии, вакуолизации стволовых клеток костного мозга и патологии экзокринной функции поджелудочной железы.
Всего было описано порядка 60 случаев синдром Пирсона у мальчиков и девочек. Гематологические симптомы манифестируют в детском возрасте, хотя описано и несколько случаев у новорожденных.
Этиология и патогенез [ править ]
Хотя наследование от матери было описано для синдрома Пирсона, заболевание, как правило, носит спорадический характер. В целом синдром Пирсона – это митохондриальная цитопатия, обусловленная делециями митохондриальной ДНК, что является важным диагностическим признаком патологии. Данные делеции приводят к нарушениям митохондриальной функции дыхательной цепи. Случайное распределение митохондриальной ДНК при делении клеток приводит к присутствию в одной клетке как нормальной, так и мутировавшей ДНК. Это сосуществование, названное гетероплазмией, объясняет высокую изменчивость в клинической экспрессии как между разными пациентами, так и между различными органами пораженного субъекта. Патологические проявления возникают, когда некоторый критический уровень мутантной ДНК накапливается в конкретной ткани.
Клинические проявления [ править ]
Клинические проявления синдрома Пирсона включают макроцитарную сидеробластную анемию, иногда сопровождаемую тяжелой нейтропенией или тромбоцитопенией.
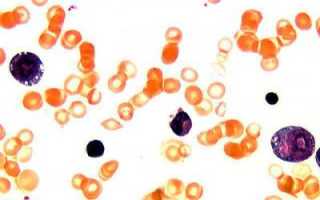
Вакуоли в клетках-предшественниках гранулоцитов и эритробластах, обнаруживаемые на миелограмме, также характерны для данного синдрома. Окраска по Перлзу обнаруживает наличие кольцевидных сидеробластов.
Кроме того проявляется фиброзная экзокринная дисфункция поджелудочной железы с симптомами мальабсорбции и поносом и нарушениями окислительного фосфорилирования, приводящим к лактатемии и увеличением соотношения лактат/пируват. Другие органы также могут быть затронуты, либо одновременно, либо по ходу течения заболевания: часто это почечная тубулопатия с аминоацидурией, гепатомегалия, сиптомы цитолиза и холестаза, эндокринные нарушения, нервно-мышечные расстройства, а в некоторых случаях поражения сердца и атрофия селезенки.
Наследственная сидеробластная анемия: Диагностика [ править ]
Дифференциальный диагноз [ править ]
Наследственная сидеробластная анемия: Лечение [ править ]
Не существует специфического лечения синдрома Пирсона. Терапия является симптоматической и включает в себя лечение инфекций и метаболических нарушений, переливание крови в случае развития тяжелой анемии (иногда совместно с назначением эритропоэтина), назначение ферментов полжелудочной железы и коррекция эндокринных нарушений.
Смерть часто происходит в возрасте до трех лет в результате сепсиса, метаболический криза с лактоацидозом или гепатоцеллюлярной недостаточности. Пациенты, которые выживают в младенческом возрасте, как правило, подвергаются фенотипической эволюции: гематологические проявления исчезают сами по себе, в то время появляются, либо прогрессируют неврологические расстройства и миопатия. У некоторых пациентов развивается типичный синдром Кернс-Сейра с офтальмоплегией, атаксией, пигментным ретинитом, дефектами проводимости и миопатией.
Профилактика [ править ]
Прочее [ править ]
Аутосомно-рецессивная сидеробластная анемия со взрослым началом
Синонимы: GLRX5-связанная сидеробластная анемия
Аутосомно-рецессивная сидеробластная анемия со взрослым началом – очень редкая несиндромальная аутосомно-рецессивная пиридоксин-рефрактерная сидеробластная анемия в результате дефекта сплайсирования глутаредоксина-5 (GLRX5). GLRX5-связанная сидеробластная анемия описана у одного взрослого пациента с микроцитарной гипохромной анемией, перегрузкой печени железом и диабетом 2 типа .
Пирсона синдром: симптомы и лечение
Синдром Пирсона – это очень редкое генетическое заболевание, которое проявляется еще в младенчестве и в большинстве случаев ведет к раннему летальному исходу.
История открытия
Другое название синдрома Пирсона – врожденная сидеробластная анемия с экзокринной недостаточностью поджелудочной железы. Болезнь названа в честь ученого, впервые ее описавшего в 1979 году – Н. А. Пирсона. Синдром был распознан благодаря длительным наблюдениям за четырьмя детьми со схожими симптомами: у них наблюдалась сидеробластная анемия, которая не поддавалась стандартному лечению, недостаточность экзокринной функции поджелудочной железы и патология клеток костного мозга.
Сначала детям ставили другой диагноз – синдром Швахмана (врожденная гипоплазия поджелудочной железы). Но после исследования крови и костного мозга были выявлены явные отличия, что и дало повод выделить Пирсона синдром в отдельную категорию.
Причины возникновения болезни
Исследование причин заболевания заняло около десяти лет. Врачи-генетики сумели найти генетический дефект, который ведет к делению и дупликации митохондриальной ДНК.
Хотя болезнь и является генетической, обычно мутация появляется спонтанно, и больной малыш рождается у абсолютно здоровых родителей. Иногда отмечают зависимость между наличием офтальмопатии у матери и развитием синдрома Пирсона у ее ребенка.
Дефекты ДНК удается выявить в костном мозге, ациноцитах поджелудочной железы, а также в органах, которые не являются главными мишенями болезни – почках, сердечной мышце, гепатоцитах. С другой стороны, у некоторых пациентов при наличии типичной клинической и лабораторной картины так и не удается зарегистрировать изменения в митохондриальной ДНК.
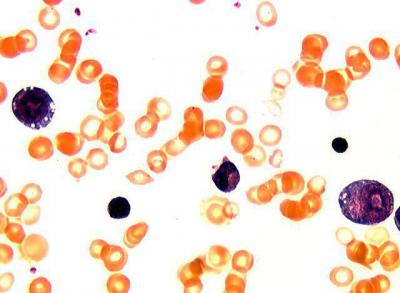
У больных детей происходит накопление железа в печени, склероз клубочков почек, образование кист. В некоторых случаях развивается фиброз миокарда, что приводит к сердечной недостаточности.
Поджелудочная железа выделяет недостаточное количество липазы, амилазы и бикарбонатов у всех пациентов с болезнью Пирсона. Синдром проявляется атрофией ткани железы и ее последующим фиброзом.
Методы диагностики
С уверенностью поставить диагноз могут только врачи-генетики после исследования митохондриальной ДНК. Также важную роль играет обычный анализ периферической крови: выявляют макроцитарную анемию тяжелой степени, нейтропению и тромбоцитопению. Примечательным является отсутствие эффекта от лечения анемии “Цианкобаламином” и препаратами железа.
Благодаря пункции костного мозга можно увидеть уменьшение общего количества клеток, наличие вакуолей в эритробластах и появление кольцевидных сидеробластов.

Симптомы болезни
Уже с первых дней жизни ребенка можно заподозрить синдром Пирсона. Симптомы болезни дебютируют у младенцев в виде злокачественной анемии и инсулинзависимого сахарного диабета. Наблюдаются бледность кожи, сонливость, вялость, диарея, периодическая рвота, ребенок плохо прибавляет в весе. Пища почти не усваивается, характерна стеаторея. Возникают симптомы сахарного диабета, повышается уровень глюкозы в крови, и появляется склонность к ацидозу. Возможно развитие печеночной, почечной и сердечной недостаточности.
Иногда кроме анемии возникает панцитопения (дефицит не только эритроцитов, но и тромбоцитов и лейкоцитов), что будет проявляться склонностью к кровотечениям и частому присоединению инфекций.

Лечение и прогноз
К сожалению, врачи до сих пор не знают, как победить синдром Пирсона. Лечение его неспецифическое и дает лишь кратковременные результаты.
Анемия не поддается стандартной терапии и требует частого переливания крови. Для улучшения функции поджелудочной железы назначают прием ферментов, а для коррекции метаболических нарушений – инфузионную терапию. В редких случаях проводят трансплантацию костного мозга.

Пирсона синдром имеет неблагоприятный прогноз: дети отстают в физическом развитии, большинство погибает до двух лет. В единичных случаях пациенты живут дольше благодаря эффективной поддерживающей терапии, однако в более старшем возрасте болезнь приводит к мышечной атрофии, характерной для синдрома Кернса-Сейра.
Тяжесть течения болезни во многом зависит от степени поражения ДНК.
Агенезия, аплазия и гипоплазия поджелудочной железы (Q45.0)
Версия: Справочник заболеваний MedElement

27-я международная выставка “Здравоохранение”
13-15 мая, Алматы, Атакент
Получить бесплатный билет

Выставка “Здравоохранение”
13-15 мая, Алматы, Атакент
Получить бесплатный билет
Общая информация
Краткое описание
Гипоплазия (недоразвитие) поджелудочной железы может быть тотальной (значительное уменьшение размеров органа с сохранением всех его анатомических отделов) и частичной (имеется только головка ПЖ, а тело и хвост отсутствуют).
Гипоплазия может представлять собой изолированный порок или выступать одним из проявлений сложных сочетанных пороков развития не только органов желудочно-кишечного тракта, но и органов других систем (синдромы Швахмана, Кларка-Хэдвилда (см. подпункт K86.8), синдром Йохансона-Близзарда, врожденная сидеробластная анемия с экзокринной недостаточностью).
Среди перечисленных пороков необходимо отдельно отметить синдром Йохансона-Близзарда, поскольку расстройства экболической (ферментообразующей) функции ПЖ при нем являются доминирующими.
Период протекания
Минимальный период протекания (дней): 1
Максимальный период протекания (дней): не указан

– Профессиональные медицинские справочники. Стандарты лечения
– Коммуникация с пациентами: вопросы, отзывы, запись на прием
Скачать приложение для ANDROID

– Профессиональные медицинские справочники
– Коммуникация с пациентами: вопросы, отзывы, запись на прием
Скачать приложение для ANDROID
Классификация
Врожденные гипoплазии поджелудочной железы у детей
1. Тотальная гипoплазия органа:
1.1 Недоразвитие ацинозной и островковой ткани всех отделов железы/
1.2 Недоразвитие ацинозной и островковой ткани в пределах отделов, формирующихся из самостоятельных эмбриональных зачатков:
– дорсальной части;
– вентральной части.
2. Парциальная гипoплазия внешнесекреторного аппарата ПЖ:
2.1 Селективные дефициты панкреатических ферментов:
2.1.1 Изолированные:
– избирательная недостаточность трипсиногена;
– избирательная недостаточность панкреатической липазы;
– постоянное отсутствие панкреатической амилазы.
2.1.2 Сочетанные:
– сочетанная недостаточность панкреатических протеолитических ферментов и липазы;
– сочетанная недостаточность трипсина и панкреатической амилазы.
2.2 Врожденная липоматозная гипoплазия:
2.2.1 Без сопутствующих гематологических нарушений.
2.2.2 В сочетании с гематологическими проявлениями:
– синдром Shwachman-Bodian;
– синдром Burke;
– синдром Pearson-Stoddard.
2.3 Гипоплазии ПЖ в сочетании с множественными пороками развития других органов (хромосомные, генные, мультифакториальные).
Этиология и патогенез
Эпидемиология
Клиническая картина
Cимптомы, течение
Основные клинические проявления изолированной гипоплазии поджелудочной железы (как тотальной, так и частичной):
1. Врожденный сахарный диабет.
2. Признаки внешнесекреторной недостаточности (выраженная стеаторея), типичный абдоминальный панкреатический болевой синдром.
При гипоплазии ПЖ как одном из проявлений сочетанного порока развития наблюдаются симптомы, типичные для поражения других органов и систем.
4. Врожденная сидеробластная анемия. Проявления: ацинарная ткань ПЖ атрофирована, фиброзная ткань изменена. В результате наблюдается резкое снижение секреции панкреатических ферментов и бикарбонатов, развитие экзокринной недостаточности.
Наиболее тяжелая форма среди внесиндромных тотальных гипоплазий ПЖ – н едоразвитие ацинозной и островковой ткани в пределах отделов, формирующихся из самостоятельных эмбриональных зачатков дорсальной части. Она характеризуется сочетанием расстройств инкреторный (инсулиновой и глюкагоновой) и экзокринной (экболической) функций органа.
Данный вариант гипоплазий ПЖ отличается особой тяжестью, связанной с тем, что на первый план выступают расстройства углеводного обмена. На фоне быстро прогрессирующих диабетогенных метаболических сдвигов нарушения кишечного пищеварения и всасывания менее заметны или вообще не успевают развиться. Продлить жизнь ребенка возможно только при помощи рано начатого лечения инсулином и панкреатическими ферментами.
Недоразвитие только одного из двух эмбриональных зачатков ПЖ имеет более легкое течение, не сопровождается ранним дефицитом инсулина и панкреатических ферментов и может проявиться в зрелом возрасте. Описан случай (G. Lechner и R. Reag) длительной (в течение 18 месяцев) дуоденальной непроходимости у мужчины 26 лет, у которого ранее был установлен сахарный диабет. при проведении операции у него была обнаружена гипоплазия дорсального эмбрионального зачатка ПЖ с отсутствием шейки, тела, крючковидного отростка и большей части головки.
Известны также такие селективные дефициты, как врожденная постоянная недостаточность панкреатической амилазы и сочетанные дефициты панкреатических протеолитических энзимов и липазы, трипсина и панкреатической амилазы. Концентрация натрия и хлора в поте при всех перечисленных селективных дефицитах не повышается. Современные методы гистологического исследования биоптатов ПЖ структуральных отклонений в секреторных клетках не выявляют.
При синдромах Швахмана и Бурке липоматозная гипоплазия внешнесекреторного отдела ПЖ в обязательном порядке сочетается с гранулоцитопенией, первичный характер которой не вызывает сомнений.
Непостоянные признаки – отставание в росте, а также наличие метафизарных дизостозов и других нарушений со стороны скелета.
Первые клинические проявления (начинаются в первые дни или недели жизни ребенка) – расстройство стула, стеаторея.
Проявления:
1. Развивается гипотрофия Гипотрофия – расстройство питания, характеризующееся различной степенью дефицита массы тела
, обнаруживаются признаки поливитаминной недостаточности.
2. Живот увеличивается в размерах .