Основные клинические проявления хронического миелолейкоза. Хронический миелолейкоз: патогенез и лечение. Наиболее встречаемые виды миелолейкоза
Хронический миелолейкоз

Хронический миелолейкоз – это злокачественное миелопролиферативное заболевание, характеризующееся преимущественным поражением гранулоцитарного ростка. Может долгое время протекать бессимптомно. Проявляется склонностью к субфебрилитету, ощущением полноты в животе, частыми инфекциями и увеличением селезенки. Наблюдаются анемия и изменение уровня тромбоцитов, сопровождающиеся слабостью, бледностью и повышенной кровоточивостью. На заключительной стадии развиваются лихорадка, лимфоаденопатия и кожная сыпь. Диагноз устанавливается с учетом анамнеза, клинической картины и данных лабораторных исследований. Лечение – химиотерапия, радиотерапия, пересадка костного мозга.
МКБ-10

Общие сведения
Хронический миелолейкоз – онкологическое заболевание, возникающее в результате хромосомной мутации с поражением полипотентных стволовых клеток и последующей неконтролируемой пролиферацией зрелых гранулоцитов. Составляет 15% от общего количества гемобластозов у взрослых и 9% от общего числа лейкозов во всех возрастных группах. Обычно развивается после 30 лет, пик заболеваемости хроническим миелолейкозом приходится на возраст 45-55 лет. Дети до 10 лет страдают исключительно редко.
Хронический миелолейкоз одинаково распространен у женщин и у мужчин. Из-за бессимптомного или малосимптомного течения может становиться случайной находкой при исследовании анализа крови, взятого в связи с другим заболеванием или во время профилактического осмотра. У части больных хронический миелолейкоз выявляется на заключительных стадиях, что ограничивает возможности терапии и ухудшает показатели выживаемости. Лечение проводят специалисты в области онкологии и гематологии.
Причины
Хронический миелолейкоз считается первым заболеванием, при котором достоверно установлена связь между развитием патологии и определенным генетическим нарушением. В 95% случаев подтвержденной причиной хронического миелолейкоза является хромосомная транслокация, известная как «филадельфийская хромосома». Суть транслокации заключается во взаимной замене участков 9 и 22 хромосом. В результате такой замены формируется устойчивая открытая рамка считывания. Образование рамки вызывает ускорение деления клеток и подавляет механизм восстановления ДНК, что увеличивает вероятность возникновения других генетических аномалий.
В числе возможных факторов, способствующих появлению филадельфийской хромосомы у больных хроническим миелолейкозом, называют ионизирующее облучение и контакт с некоторыми химическими соединениями.
Патогенез
Итогом мутации становится усиленная пролиферация полипотентных стволовых клеток. При хроническом миелолейкозе пролиферируют преимущественно зрелые гранулоциты, но аномальный клон включает в себя и другие клетки крови: эритроциты, моноциты, мегакариоциты, реже – В- и Т-лифоциты. Обычные гемопоэтические клетки при этом не исчезают и после подавления аномального клона могут служить основой для нормальной пролиферации кровяных клеток. Для хронического миелолейкоза характерно стадийное течение.
- При первой, хронической (неактивной) фазе отмечается постепенное усугубление патологических изменений при сохранении удовлетворительного общего состояния.
- Во второй фазе хронического миелолейкоза – фазе акселерации изменения становятся явными, развиваются прогрессирующие анемия и тромбоцитопения.
- Заключительной стадией хронического миелолейкоза является бластный криз, сопровождающийся быстрой экстрамедуллярной пролиферацией бластных клеток.
Источником бластов становятся лимфатические узлы, кости, кожа, ЦНС и т. д. В фазе бластного криза состояние больного хроническим миелолейкозом резко ухудшается, развиваются тяжелые осложнения, завершающиеся гибелью больного. У некоторых пациентов фаза акселерации отсутствует, хроническая фаза сразу сменяется бластным кризом.
Симптомы хронического миелолейкоза
Клиническая картина определяется стадией заболевания. Хроническая фаза в среднем продолжается 2-3 года, в некоторых случаях – до 10 лет. Для этой фазы хронического миелолейкоза характерно бессимптомное течение или постепенное появление «легких» симптомов: слабости, некоторого недомогания, снижения трудоспособности и чувства переполнения живота. При объективном осмотре больного хроническим миелолейкозом может обнаруживаться увеличение селезенки. По анализам крови выявляется повышение количества гранулоцитов до 50-200 тыс./мкл при бессимптомном течении заболевания и до 200-1000 тыс./мкл при «легких» признаках.
На начальных стадиях хронического миелолейкоза возможно некоторое снижение уровня гемоглобина. В последующем развивается нормохромная нормоцитарная анемия. При исследовании мазка крови пациентов с хроническим миелолейкозом отмечается преобладание молодых форм гранулоцитов: миелоцитов, промиелоцитов, миелобластов. Наблюдаются отклонения от нормального уровня зернистости в ту или иную сторону (обильная или очень скудная). Цитоплазма клеток незрелая, базофильная. Определяется анизоцитоз. При отсутствии лечения хроническая фаза переходит в фазу акселерации.
О начале фазы акселерации может свидетельствовать как изменение лабораторных показателей, так и ухудшение состояния пациентов. Возможно нарастание слабости, увеличение печени и прогрессирующее увеличение селезенки. У больных хроническим миелолейкозом выявляются клинические признаки анемии и тромбоцитопении или тробоцитоза: бледность, быстрая утомляемость, головокружения, петехии, кровоизлияния, повышенная кровоточивость. Несмотря на проводимое лечение, в крови пациентов с хроническим миелолейкозом постепенно увеличивается количество лейкоцитов. При этом отмечается возрастание уровня метамиелоцитов и миелоцитов, возможно появление единичных бластных клеток.
Бластный криз сопровождается резким ухудшением состояния больного хроническим миелолейкозом. Возникают новые хромосомные аномалии, моноклоновое новообразование трансформируется в поликлоновое. Отмечается нарастание клеточного атипизма при угнетении нормальных ростков кроветворения. Наблюдаются ярко выраженные анемия и тромбоцитопения. Суммарное количество бластов и промиелоцитов в периферической крови составляет более 30%, в костном мозге – более 50%. Пациенты с хроническим миелолейкозом теряют вес и аппетит. Возникают экстрамедуллярные очаги незрелых клеток (хлоромы). Развиваются кровотечения и тяжелые инфекционные осложнения.
Диагностика
Диагноз устанавливается на основании клинической картины и результатов лабораторных исследований. Первое подозрение на хронический миелолейкоз часто возникает при повышении уровня гранулоцитов в общем анализе крови, назначенном в порядке профилактического осмотра или обследования в связи с другим заболеванием. Для уточнения диагноза могут использоваться данные гистологического исследования материала, полученного при стернальной пункции костного мозга, однако окончательный диагноз «хронический миелолейкоз» выставляется при выявлении филадельфийской хромосомы при помощи ПЦР, флюоресцентной гибридизации или цитогенетического исследования.
Вопрос о возможности постановки диагноза хронический миелолейкоз при отсутствии филадельфийской хромосомы остается дискутабельным. Многие исследователи считают, что подобные случаи могут объясняться комплексными хромосомными нарушениями, из-за которых выявление данной транслокации становится затруднительным. В ряде случаев филадельфийскую хромосому можно обнаружить при использовании ПЦР с обратной транскрипцией. При отрицательных результатах исследования и нетипичном течении заболевания обычно говорят не о хроническом миелолейкозе, а о недифференцированном миелопролиферативном/миелодиспластическом расстройстве.
Лечение хронического миелолейкоза
Тактику лечения определяют в зависимости от фазы заболевания и выраженности клинических проявлений. В хронической фазе при бессимптомном течении и слабо выраженных лабораторных изменениях ограничиваются общеукрепляющими мероприятиями. Больным хроническим миелолейкозом рекомендуют соблюдать режим труда и отдыха, принимать пищу, богатую витаминами и т. д. Лечение может включать:
- Монохимиотерапию. При повышении уровня лейкоцитов используют бусульфан. После нормализации лабораторных показателей и уменьшения селезенки пациентам с хроническим миелолейкозом назначают поддерживающую терапию или курсовое лечение бусульфаном. При бластных кризах осуществляют лечение гидроксикарбамидом.
- Радиотерапию. Облучение обычно используют при лейкоцитозе в сочетании со спленомегалией. При снижении уровня лейкоцитов делают паузу продолжительностью не менее месяца, а затем переходят на поддерживающую терапию бусульфаном. Радиотерапию также назначают при хлоромах.
- Полихимиотерапию. В прогрессирующей фазе хронического миелолейкоза возможно использование одного химиопрепарата или полихимиотерапии. Применяют митобронитол, гексафосфамид или хлорэтиламиноурацил. Как и в хронической фазе, проводят интенсивную терапию до стабилизации лабораторных показателей, в последующем переходят на поддерживающие дозы. Курсы полиохимиотерапии при хроническом миелолейкозе повторяют 3-4 раза в год.
- Гемокоррекцию. При неэффективности терапии используют лейкоцитаферез. При выраженной тромбоцитопении, анемии выполняют переливания тромбоконцентрата и эритроцитарной массы.
- ТКМ.Пересадку костного мозга проводят в первой фазе хронического миелолейкоза. Продолжительной ремиссии удается достичь у 70% пациентов.
- Удаление селезенки. При наличии показаний осуществляют спленэктомию. Экстренная спленэктомия показана при разрыве или угрозе разрыва селезенки, плановая – при гемолитических кризах, «блуждающей» селезенке, рецидивирующих периспленитах и резко выраженной спленомегалии, сопровождающейся нарушением функций органов брюшной полости.
Прогноз
Прогноз при хроническом миелолейкозе зависит от множества факторов, определяющим из которых является момент начала лечения (в хронической фазе, фазе активации или в период бластного криза). В качестве неблагоприятных прогностических признаков хронического миелолейкоза рассматривают значительное увеличение печени и селезенки (печень выступает из-под края реберной дуги на 6 и более см, селезенка – на 15 и более см), лейкоцитоз свыше 100×10 9 /л, тромбоцитопению менее 150×10 9 /л, тромбоцитоз более 500х10 9 /л, повышение уровня бластных клеток в периферической крови до 1% и более, повышение суммарного уровня промиелоцитов и бластных клеток в периферической крови до 30% и более.
Вероятность неблагоприятного исхода при хроническом миелолейкозе возрастает по мере увеличения количества признаков. Причиной гибели становятся инфекционные осложнения или тяжелые геморрагии. Средняя продолжительность жизни пациентов с хроническим миелолейкозом составляет 2,5 года, однако при своевременном начале терапии и благоприятном течении заболевания этот показатель может увеличиваться до нескольких десятков лет.
Хронический миелолейкоз: этиология, патогенез, клиника, диагностика, принципы лечения
Хронический миелолейкоз (ХМЛ) – миелоидная опухоль, развивающаяся из полипотентной клетки-предшественницы, пролиферация и дифференцировка которой приводит к расширению ростков кроветворения, представленных преимущественно зрелыми и промежуточными формами. Закономерным исходом ХМЛ является бластный криз.
Эпидемиология: 20% всех лейкозов, на 3-ем месте по распространенности после острых лейкозов и ХЛЛ.
Клеточный субстрат ХМЛ: преимущественно гранулоциты (в основном нейтрофилы).
Этиология ХМЛ – достоверно доказана роль в возникновении ХМЛ:
а) ионизирующего излучения (пострадавшие от бомбардировки Хиросимы и Нагасаки; больные спондилоартритом, получавшие рентгенотерапию)
б) химических агентов (бензол и др. органические растворители)
1. Перенос большей части длинного плеча хромосомы 22 на длинное плечо хромосомы 9, а малой терминальной части длинного плеча 9 хромосомы на хромосому 22 с образованием Рh-хромосомы («филадельфийской» – диагностический признак для ХМЛ)
2. На длинном плече хромосомы 9 расположен протоонкоген ABL, кодирующий образование белка семейства тирозинпротеинкиназ; при транслокации часть протоонкогена переносится на длинное плечо хромосомы 22 в тот участок, где произошел разрыв и находится ген BCR; в результате этого слияния на хромосоме 22 образуется ген BCR-ABL, который кодирует химерный белок, имеющий более выраженную тирозинкиназную активность, чем его нормальный прототип.
3. Активация различных участков химерного гена обусловливает цепь событий, ведущих к увеличению клеточной пролиферации.
Клиника и диагностика ХМЛ:
а) начальная стадия – ХМЛ практически не выявляется (при случайном исследовании ОАК можно выявить изменения: лейкоцитоз, базофильно-эозинофильная ассоциация), клиника отсутствует
б) стадия акселерации (развернутая стадия):
– появляется быстрая утомляемость, потливость, субфебрилитет, потеря массы тела
– тяжесть и боли в левом подреберье при увеличении селезенки (она может быть даже в малом тазу); возможны инфаркты селезенки с отрыми болями
– печень слабо увеличена
– л/у практически не увеличены
– геморрагический синдром обычно отсутствует
– в легких – пневмония, связанная с лейкемической инфильтрацией и вторичной инфекцией
1) ОАК – нормохромная анемия средней степени тяжести, лейкоцитоз, увеличение базофилов и эозинофилов, снижение лимфоцитов и немного – моноцитов, присутствие всех форм нейтрофилов (миелобластов, промиелоцитов, юных, палочкоядерных, сегментоядерных, лейкемический провал отсутствует), тромбоциты умеренно снижены, СОЭ ускорено
2) миелограмма – увеличение числа мегакариоцитов, процента незрелых гранулоцитов с повышением миелоидно-эритроидного соотношения до 20-25 : 1 (в норме – 3-4 : 1); снижение ЩФ нейтрофилов менее 25 ед; Ph-хромосома в кроветворных клетка миелоидного ряда
3) БАК – увеличение витамина В12 в 10-15 раз, увеличение мочевой кислоты (гиперурикемический синдром), значительное повышение ЛДГ.
в) терминальная стадия (бластный криз) –характерно появление в клетках костного мозга дополнительных хромосомных нарушений, в период ремиссии бластного криза эти дополнительные хромосомные нарушения исчезают:
– ухудшение самочувствия, стойкое повышение температуры, истощение больного
– увеличение селезенки и в меньшей степени печени
– дистрофические изменения внутренних органов
– рефрактерность к проводимой терапии
– частые инфекционные осложнения и др. проявления острого лейкоза
1. Химиотерапия:
В лечении ХМЛ применяются ряд препаратов: а) миелосан (бусульфан, милеран) – алкилирующий препарат, использовался в лечении ХМЛ первым; б) гидроксимочевина (литалир) – ингибитор рибонуклеотидазы – фермента, необходимого для синтеза ДНК; в) α-интерферон – позволяет получить гематологическую, цитогенетическую ремиссию, понижает уровни ЛДГ и витамина В12; г) цитозар – пиримидиновый нуклеозид, активный метаболит которого угнетает ДНК-полимеразу, что ведет к нарушению синтеза ДНК и подавлению роста Ph-позитивных клеток; д) рубомицин-антрациклин
В настоящее время фазу акселерации и терминальную фазу применяют схему 3+7 (рубомицин-антрациклин + цитозар) каждые 1,5-2 месяца.
Новые препараты в терапии ХМЛ:
а) гомохаррингтонин – синтетический аналог китайского растительного алкалоида
б) децитабин – ингибитор гиперметилирования в клеточных циклах (данный процесс наблюдается при прогрессировании опухолей, в период бластного криза)
в) полностью транс-ретиноевая кислота (ATRA) + IFNα
г) топотекан – ингибитор фермента топоизомеразы I, необходимой для репликации ДНК
е) ингибитор мутантной тирозинкиназы Гливек (иматиниб мозилат) – соединяесь с активными центрами BCR-ABL-тирозинкиназы, он нарушает процессы взаимодействия субстратов внутри клетки, что приводит к гибели клеток, содержащих белок р210, т.е. Ph-позитивных
2. Лучевая терапия: показана при резко выраженной спленомегалии и перисплените, при экстрамедуллярных опухолевых образованиях, угрожающих жизни больного
3. При угрозе разрыва селезенки показана спленэктомия.
4. В период клинико-гематологической ремиссии показана трансплантация костного мозга.
59. Хронический лимфолейкоз: этиология, патогенез, клинические варианты, диагностика, осложнения, принципы лечения.
Хронический лимфолейкоз (ХЛЛ) – клональное лимфопролиферативное неопластическое заболевание, характеризующееся пролиферацией и увеличением в периферической крови количества зрелых лимфоцитов на фоне лимфоцитарной инфильтрации костного мозга, лимфатических узлов, селезенки и других органов.
Эпидемиология: около 30% всех лейкозов; заболеваемость 2,7 на 100 тыс.; болеют в основном пожилые; в 95% случаев клеточный субстрат представлен В-лимфоцитами, в 5% – Т-лимфоцитами
1) ретровирусы – их роль в развитии ХЛЛ сегодня уже очевидна
2) наследственная предрасположенность (хромосомные аномалии, обнаруживаются в 100% случаев; наиболее характерны трисомия 12, делеция длинного плеча 13 хромосомы, избыток генетического материала в 14 хромосоме и др.)
В настоящее время развитие ХЛЛ не ассоциируется с воздействием ионизирующего излучения, лекарств, химических веществ.
Патогенез В-клеточного ХЛЛ (как наиболее частой формы):
1. Возникновение патологического клона клеток-предшественниц, дифференцирующихся только по В-лимфоцитарному пути, вследствие воздействия вирусов или спонтанных генетических мутаций
2. Развитие патологического клона клеток по законам опухолевой прогрессии, но значительно медленнее и менее агрессивно, чем при остром лейкозе, с преобладанием зрелых лимфоцитов, накапливающихся вначале в лимфатических узлах, далее – в лимфоидных тканях, печени, селезенки, костном мозге.
3. Прогрессирующая лимфоцитарная инфильтрация костного мозга, приводящая к нарушениям гемопоэза (анемии, гранулоцитопении, тромбоцитопении), продукции иммуноглобулинов, возникновению аутоиммунных конфликтов (из-за неполноценности лимфоцитов патологического клона).
Клинические проявления и диагностика ХЛЛ:
а) начальный период:
– общее состояние удовлетворительное, жалобы на небольшую слабость, потливость, частые простудные заболевания
– небольшое увеличение л.у. (в первую очередь – шейных, затем подмышечных, остальные л.у. увеличиваются в развернутой стадии заболевания); л.у. эластично-тестотваты, безболезненны, не спаяны с кожей и между собой, не изъязвляются и не нагнаиваются
Диагностика: лейкоцитоз 10-30*10 9 /л, не имеющий тенденции к увеличению; абсолютный лимфоцитоз до 60-80%
б) период выраженных клинических проявлений:
– жалобы на резко выраженную общую слабость, снижение работоспособности, значительную потливость, особенно ночью, похудание, повышение температуры тела
– выраженная лимфаденопатия (увеличены практически все группы периферических л.у. в различной степени – от горошины до куринового яйца); л.у. эластично-тестоватые, безболезненные, не спаяны между собой и с кожей
– неспецифические поражения кожи (обострения ранее существовавших болезней – экземы, псориаза; опоясывающий герпес, иногда генерализованного характера; крапивница, нейродермит; грибковые поражения)
– проявления лейкемической инфильтрации внутренних органов:
– значительно увеличенная селезенка (но меньше, чем при ХМЛ), плотная селезенка с гладкой поверхностью, реже – увеличенная печень с загругленным плотноватым краем и гладкой поверхностью
– язвенное поражение желудка, ДПК, желудочно-кишечные кровотечения, синдром мальабсорбции (из-за лейкемической инфильтрации слизистой ЖКТ)
– очаговые (клинически не проявляются) или диффузные диссеменированные (клинически – одышка, кашель, кровохарканье, крепитация над всей поверхностью легких) инфильтраты в легких; фибринозный или экссудативный плеврит; частые инфекционно-воспалительные заболевания органов дыхания
– очаговые поражения миокарда (клинически не проявляются) или миокардиодистрофия; повышение проницаемости сосудов различного калибра
– поражение почек с умеренной протеинурией, реже микрогематурией без нарушения их функции
– приапизм у мужчин (длительная и болезненная эрекция из-за лейкемической инфильтрации кавернозных тел)
– нейролейкемия с развитием менингита, менингоэнцефалита, параличей и даже комы
1) ОАК: лейкоцитоз (50-200*10 9 /л), резкий лимфоцитоз периферической хрови (10*10 9 /л и более при норме 9 /л); в лейкоцитарной формуле лимфоцитов 80-90%, преимущественно зрелых (размеры 10-12 мкм, форма округлая или овальная, ядро расположено экцентрично, хроматин гомогенный, разделен светлыми бороздами, цитоплазма неширокая, светло-голубая); тени Боткина-Гумпрехта (полуразрушенные ядра лимфоцитов); нормохромная нормоцитарная анемия; тромбоцитопения; увеличение СОЭ
2) миелограмма: выраженная лимфоидная инфильтрация (более 30% лимфоцитов от общего числа миелоцитов), значительное уменьшение гранулоцитов
3) пункция л.у. – проводится в трудных дифференциально-диагностических случаях; пунктат состоит из преимущественно зрелых малых лимфоцитов
4) пункция селезенки – выполняется при алейкемическом лимфолейкозе, протекающем без увеличения
л.у. и без четких морфологических проявлений ХЛЛ в стернальном пунктате), главным образом при доминировании в клинической картине спленомегалии; характерно значительное увеличение количества лимфоцитов и пролимоцитов
5) ОАМ: протеинурия, нерезко выраженная микрогематурия
6) БАК: гипогаммаглобулинемия, которая усугубляется по мере прогрессирования процесса; гипоальбуминемия;
изменения ферментов печени
7) иммунологический анализ крови и иммунотипирование: определение В- или Т-клеточной формы ХЛЛ; снижение содержания в крови IgG и особенно IgA и IgM; цитогенетическое и цитохимическое исследование лимфоцитов
8) методы инструментальной диагностики (УЗИ, КТ и др.) для выявления поражения внутренних органов.
в) терминальная стадия:
– резко прогрессирующее ухудшение общего состояния больных, истощение, выраженная интоксикация, исчезновение аппетита, высокая температура тела
– развитие тяжелых осложнений (различных инфекционно-воспалительных процессов, почечной недостаточности, нейролейкемии, кардиомиопатии и сердечной недостаточности, геморрагических кровотечений)
1) инфекционно-воспалительные процессы (особенно характерен генерализованный герпес), частые флегмоны (в т.ч. и от инъекций), присоединение внутрибольных инфекций, туберкулеза
2) ателектазы легких, нарушения вентиляции (при гиперплазии лимфатических фолликулов бронхиального дерева и инфильтрации опухолевыми клетками самой легочной ткани), плевриты
3) ХПН (при инфильтрации паренхимы почек)
4) синдром цитолиза (гемолиз, анемия, ретикулоцитоз, тромбоцитопения и тяжелые геморрагии)
5) кардиомиопатия и развитие сердечной недостаточности (при инфильтрации миокарда)
Основные клинические варианты ХЛЛ: а) доброкачественная; б) прогрессирующая; в) спленомегалическая; г) абдоминальная; д) опухолевая; е) костномозговая; ж) пролимфоцитарная.
Для определения показаний к началу лечения выделяют клинические стадии ХЛЛ: 0 стадия – Т-лимфоцитоз; 1 стадия – Т-лимфоцитоз и лимфаденопатия, 2 стадия – сплено- или гепатомегалия, 3 стадия – аутоиммунная гемолитическая анемия, 4 стадия – аутоиммунная тромбоцитопения.
1. На ранней стадии болезни при стабильном лейкозе лечение не проводят, показано лишь наблюдение и периодический контроль анализов крови (терапия «ждать и наблюдать»); показания к началу лечения: ухудшение общего состояния (появление лихорадки, потливости, похудания и др.) с увеличением л.у. и внутренних органов; нарастание лейкоцитоза выше 50*10 9 /л; появление аутоиммунных феноменов; учащение и тяжесть инфекционных осложнений.
2. Первоначальная цитостатическая терапия:
а) при лейкоцитозе и умеренной лимфаденопатии: лейкеран (хлорбутин) 4-10 мг 1 раз/сут, затем терапия поддержания 4-8 мг через день
б) при лейкоцитозе и выраженной лимфаденопатии: циклофосфан (эндоксан) 200-400 мг внутрь 1 раз/сут, затем прерывистая терапия 200-300-400 мг 1 раз/сут через день 10 дней, после двухнедельного перерыва – повторить курс.
3. При неэффективности монохимиотерапии – полихимиотерапия по следующим программам: СНОР – циклофосфамид, винкристин, адриамицин, преднизолон; СОР – циклофосфамид, винкристин, преднизолон; САР – циклофосфамид, адриамицин, преднизолон; М2 – циклофосфамид, кармустин, винкристин, мелфалан, преднизолон и др.
4. При резистентности к цитостатикам: лимфоцитоферез + введение гамма-иммуноглобулина.
5. Дистанционная гамма-терапия показана при резком увеличении отдельных групп л.у. и селезенки и при выраженной генерализованной лимфаденопатии.
Новые перспективные препараты для лечения ХЛЛ: гемцитабин; кладрибин; мабтера (ритуксимаб – химерные антитела против поверхностного В-клеточного CD20); антитела Campath-1H (анти-CD52).
Клинические проявления хронического миелолейкоза
В течении этого заболевания выделяют три стадии:
· 1 стадия – начальная. Характеризуется миелоидной пролиферацией только костного мозга и небольшими изменениями в периферической крови без явлений интоксикации.
· 2 стадия – развернутая. Проявляется выраженными клинико-гематологическими проявлениями (миелоидная пролиферация костного мозга и других внутренних органов, выраженные изменения в периферической крови, признаки интоксикации).
· 3 стадия – терминальная. Соответствует развитию поликлоновой опухоли. Характеризуется развитием абсолютной рефрактерности к проводимой цитостатической терапии, значительным увеличением печени и, особенно, селезенки, общим истощением, дистрофическими изменениям внутренних органов, подавлением других ростков кроветворения (выраженная анемия, тромбоцитопения, геморрагический синдром и др.).
Начальная стадия заболевания протекает с маловыраженными клиническими признаками и сопровождается выраженным астеническим синдромом: немотивированная общая слабость, быстрая утомляемость, повышенная потливость, потеря аппетита, нарушение сна, по которым трудно заподозрить системное заболевание крови. Лимфоузлы, печень и селезенка не увеличены. Однако уже в эту стадию при проведении традиционного анализа крови, помимо зрелых форм нейтрофилов, могут выявляться миелоциты, метамиелоциты, промиелоциты и миелобласты, что может стать решающим фактором в диагностике хронического миелолейкоза. Анемия на раннем этапе развития заболевания не характерна. Замечено, что уже в начальную стадию хронического миелолейкоза выявляется базофильно-эозинофильная ассоциация, т.е. одновременное увеличение базофилов (более 2%) и эозинофилов (свыше 9%) в периферической крови.
Развернутая стадия хронического миелолейкоза, наряду с клиническим признаками начального периода, характеризуется дальнейшим развитием миелопролиферативного синдрома, который выходит за рамки костного мозга и распространяется на лимфоузлы, печень, селезенку. Кроме того, очаги миелоидной пролиферации появляются в других внутренних органах. Нарастают признаки анемии и интоксикации.
Больные жалуются на выраженную общую слабость, головокружение, шум в ушах, потемнение в глазах, обусловленные анемическим синдромом. Отмечается периодическое повышение температуры, обусловленное повышенным клеточным распадом, оссалгии, артралгии, боли в левом и правом подреберьях.
 При общем осмотре в эту стадию отмечается бледность кожи и слизистых оболочек, довольно часто наблюдается лимфаденопатия, проявляющаяся увеличением и уплотнением шейных, подмышечных и паховых лимоузлов. Пункция или биопсия выявляют лейкемическую миелоидную инфильтрацию лимфоузлов миелобластами, промиелоцитами, миелоцитами и, даже, сегментоядерными нейтрофилами.
При общем осмотре в эту стадию отмечается бледность кожи и слизистых оболочек, довольно часто наблюдается лимфаденопатия, проявляющаяся увеличением и уплотнением шейных, подмышечных и паховых лимоузлов. Пункция или биопсия выявляют лейкемическую миелоидную инфильтрацию лимфоузлов миелобластами, промиелоцитами, миелоцитами и, даже, сегментоядерными нейтрофилами.
Увеличение селезенки (спленомегалия) – наиболее характерный признак хронического миелолейкоза. Она иногда достигает очень больших размеров, занимая большую часть брюшной полости, достигая нижним полюсом малого таза. Селезенка плотная, что обусловлено,
Рис.74. Увеличение печени
и селезенки при хроническом
помимо лейкемической инфильтрации, фиброзом и рубцовыми изменениями в ее капсуле. Одновременно может увеличиваться и печень (рис.74).
При отсеве очагов миелоидной гиперплазии в легкие и плевру появляется сухой кашель, одышка. При объективном обследовании могут выявляться признаки экссудативного плеврита.
Лейкемическое поражение миокарда сопровождается колющими или ноющими болями в области сердца, сердцебиением, перебоями в работе сердца, одышкой, отеками на нижних конечностях. Отмечается акроцианоз, перкуторно – расширение границ относительной сердечной тупости влево, при аускультации – ослабление 1 тона на верхушке сердца. На ЭКГ – снижение вольтажа зубцов и особенно зубца Т, который довольно часто становится отрицательным.
При анализе крови у большинства больных общее количество лейкоцитов увеличивается до 150-200х10 9 /л, из них гранулоциты составляют 95% и больше со сдвигом лейкоцитарной формулы до промиелоцитов, наличием всех промежуточных форм созревающих лейкоцитов нейтрофильного ряда. В эту же стадию отмечается снижение эритроцитов и гемоглобина, СОЭ может увеличиваться до 50-60 мм/час.
В костномозговом пунктате резко преобладают миелоидные клетки (миелобласты, промиелоциты, миелоциты).
Терминальная стадия хронического миелолейкоза характеризуется прогрессирующим похуданием, общей адинамией в сочетании с усилением болей в костях, значительным подъемом температуры тела на фоне возникновения бластных кризов, свидетельствующих об озлокачествлении патологического кроветворения. Бластный криз характеризуется нарастанием бластов в костном мозге и крови (до 5-10%). Они представлены главным образом миелобластами, хотя возможны варианты с миеломонобластами, монобластами, эритробластами или мегакариобластами, что говорит о переходе процесса в поликлоновую стадию. Состояние больных резко ухудшается, нарастает интоксикация.
Кожа серовато-воскового цвета, иногда с землистым оттенком, дряблая, нередко покрыта липким потом, особенно при повышении температуры тела. Иногда на коже лица и конечностей выявляются очаги некроза. Одно из проявлений терминальной стадии – появление лейкемидов в коже. Они выглядят слегка приподнимающимися над поверхностью папулами розовато-песочного или красновато-коричневого цвета, плотными наощупь, безболезненными.
Очень важным признаком терминальной стадии хронического миелолейкоза является выявление в биоптате лимфоузла опухолевых клеток типа сарком, в которых есть Ph-хромосома.
В ряде случаев начало терминальной стадии сопровождается довольно быстрым увеличением селезенки с развитием в ней инфарктов и перисплинита. Над ней выслушивается шум трения брюшины.
Важным и довольно раним признаком терминальной стадии хронического миелолейкоза является развитие рефрактерности к проводимому лечению. Такими же признаками является нарастающая анемия, геморрагические проявления (петехии, экхимозы, кровотечения, которые довольно часто приводят к летальному исходу).
Причинами смерти больных хроническим миелолейкозом могут стать также сердечная, печеночная или почечная недостаточности, нарастающая кахексия и различные инфекционные осложнения (гнойные пневмонии, плевриты, бронхиты, гнойные поражения кожи и подкожной клетчатки).
Хронический лимфолейкоз (ХЛЛ)
Это заболевание относится к лимфопролиферативным опухолям, происходящим из Т- и В-лимфоцитов, и представляет собой относительно доброкачественную опухоль, основной морфологический субстрат которой составляют зрелые и созревающие лимфоциты, но функционально не полноценные, так как не выполняют свою защитную функцию.
В странах Европы и Северной Америки в 95-98% случаев субстратом опухоли являются В-лимфоциты, в азиатских странах (Япония и Китай) преобладает Т-клеточный хронический лимфолейкоз. Болезнь характеризуется лимфатической пролиферацией лимфоузлов, костного мозга, селезенки и печени, а также других органов и систем.
Этиопатогенез хронического лимфолейкоза имеет некоторые отличительные особенности.
1. Большое значение имеет наследственно-семейная предрасположенность и нарушения иммунологической реактивности. ХЛЛ является самой частой формой лейкоза у кровных родственников.
2. Как правило отсутствует связь с мутагенными факторами, в частности с радиацией, химическими агентами, вирусом Эпштейна-Барр и др.
В то же время, согласно данным VIII Международного рабочего совещания по ХЛЛ (1999г.), у 90% больных выявляются различные хромосомные аберрации: у 55% больных выявляются дефекты в хромосоме 13, у 18% – в хромосоме 11, у 7% – в хромосоме 17 и др.
3. Болезнь развивается в определенных этнических группах, чаще болеют пожилые мужчины.
Около 70% пациентов заболевают между 50 и 70 годами. Средний возраст к началу заболевания составляет 55 лет. В странах Азии и Африки ХЛЛ является редким заболеванием. В Японии, например, на всю страну регистрируется не боле одного нового случая ХЛЛ в год. Если среди белого населения в Северной Америке на долю ХЛЛ приходится 9% от злокачественных заболеваний, то среди черного – только 0,7%. Отмечается повышенная его частота среди евреев.
4. В его течении, как правило, отсутствуют признаки опухолевой прогрессии, большая редкость развития бластного криза в терминальной стадии.
5. Нет врожденного морфологического атипизма опухолевых клеток или он встречается крайне редко при злокачественном волосатоклеточном лимфолейкозе.
В течении хронического лимфолейкоза принято выделять начальный период, развернутый и терминальный. Есть и другие классификации стадий ХЛЛ. В 1981г. была разработана новая классификация стадий ХЛЛ (J. Binet с соавт), которую мы приводим ниже. Она широко распространена в Европе.
Хронический миелолейкоз: патогенез и лечение

Что такое хронический миелолейкоз?

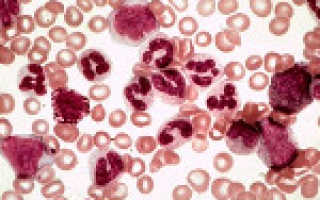
Мазок крови пациента с хроническим миелолейкозом
Хронический миелолейкоз (ХМЛ) — злокачественное новообразование кроветворной ткани, сопровождающееся прогрессирующей пролиферацией незрелых гранулоцитов. Заболевание изначально обладает вялотекущим характером, постепенно перетекая в стадию обострения с выраженной симптоматикой и образованием системных нарушений. Является одной из самых опасных и инвалидизирующих болезней.
ХМЛ — первое онкологическое заболевание, у которого определена связь между развитием канцерогенеза и мутацией в гене. Характерная аномалия основана на транслокации 9-й и 22-й хромосом, то есть участки данных хромосом меняются местами, образуя аберрантную хромосому. Выявлена мутировавшая хромосома исследователями из Филадельфии, поэтому она получила название филадельфийская или Ph-хромосома.
Причины развития

Негативное воздействие на кроветворение оказывают ядохимикаты
Заболевание известно науке с 1811 года, но до сих пор факторы, провоцирующие мутацию в гене, определить не удалось. Существует ряд причин, способствующих развитию патологии:
- радиоактивное облучение, в том числе при лучевой терапии;
- химиотерапия иных онкологических заболеваний;
- ряд генетических заболеваний, характеризующихся хромосомной аномалией (например, синдром Дауна);
- взаимодействие с химическими соединениями (нефтепродукты, пестициды).
Патогенез хронического миелолейкоза

Патогенез хронического миелолейкоза
Гибридный ген BCR-ABL 1, образованный в результате транслокации хромосом, продуцирует синтез белка BCR-ABL. Данный белок представляет собой тирозинкиназу, которая в норме способствует передаче сигнальных импульсов для роста клетки. Созданная путём мутации тирозинкиназа становится активным фактором пролиферации клеток, они начинают делиться и распространяться уже независимо от факторов роста. Происходит процесс создания клонов мутировавшей клетки.
Бесконтрольное деление сопровождается нарушением апоптоза — запрограммированной гибели клеток. Также гибридная тирозинкиназа подавляет естественные функции восстановления в молекулах ДНК, создавая предпосылки для последующих мутаций, что усугубляет патологический процесс.
Размножающиеся клетки являются незрелыми, бластными предшественниками полноценных элементов крови. Постепенно бластные клетки вытесняют функциональные эритроциты, тромбоциты и лейкоциты. Добавляются нарушения и в других хромосомах, что запускает ускоренный процесс разрушения организма в целом.
Стадии хронического миелолейкоза

Бластный криз — одна из стадий миелолейкоза
- Хроническая — 30% бластных клеток. Стадия характеризуется агрессивным характером мутировавших клеток, состояние пациента резко ухудшается. Дополнительные аномалии как в гене BCR-ABL, так и в геноме в целом, провоцируют цепь патологических реакций, которые уже практически не поддаются лечению. На этом этапе могут поражаться ткани внутренних органов, кожные покровы и слизистые оболочки, миелоидные клетки преобразовываются в саркому.
Симптомы и признаки

Признаки ХМЛ становятся заметны ближе к прогрессирующей стадии.
- Симптомы опухолевой интоксикации: снижение массы тела, быстрая утомляемость, волнообразное повышение температуры, кожный зуд, тошнота, суставные боли.
- Симптомы опухолевой пролиферации — увеличение селезёнки и печени, боль в левом подреберье, поражение кожных покровов.
- Анемический синдром — головокружение, выраженная бледность, учащённое сердцебиение, чувство нехватки воздуха.
- Геморрагический синдром — склонность к кровоточивости слизистых оболочек, сыпь в виде красных точек, длительное кровотечение при незначительных порезах.
Диагностика заболевания

Один из методов диагностики заболевания — рентгенологический
Диагностика ХМЛ включает:
- Первичный осмотр пациента с изучением анамнеза, жалоб, а также исследование при помощи пальпации размеров селезёнки и печени.
- Общий анализ крови выявляет число и характеристики форменных элементов крови.
- Биохимический анализ проводится для определения уровня билирубина, электролитов, глюкозы, ЛДГ, АСТ, АЛТ.
- Гистологическое исследование костного мозга определяет скопления бластных клеток.
- Цитогенетический анализ выявляет транслокацию хромосом.
- На 3-й стадии проводится иммунофенотипирование для идентификации бластных клеток.
- Метод генного секвенирования применяется для выявления генных мутаций.
- Проводится УЗИ внутренних органов, в первую очередь селезёнки и печени.
- Дополнительно назначают рентгенографию органов грудной клетки, ЭКГ, эхокардиографию, ИФА на маркеры различных заболеваний, коагулограмму и другие исследования.
Лечение

Основа лечения — ингибиторы тирозинкиназы
Терапия ХМЛ в настоящее время основана на применении ингибиторов тирозинкиназы. Средство I поколения иматиниб блокирует деятельность гибридной тирозинкиназы, проникая в «карман» белка BCR-ABL. Создание иматиниба совершило прорыв в лечении ХМЛ благодаря своей эффективности. Однако нередко у пациентов возникает устойчивость к препарату, что привело к созданию ингибиторов II поколения. Сочетание с другими методами лечения позволяет достичь высоких показателей в улучшении качества и продолжительности жизни.
Выбор препарата и доза определяются в зависимости от стадии ХМЛ и риска побочных эффектов. Обычно лечение начинается с приёма иматиниба в дозировке 400 мг/день при начальной стадии, 600 мг/день при последующих стадиях, затем дозу могут увеличивать или снижать. Различные аберрации в генах обусловливают низкую чувствительность к препаратам, поэтому пациенту могут менять одни ингибиторы на другие.
![]()
![]()
Трансплантация костного мозга
Если терапия не оказывает действия, рекомендуется аллогенная трансплантация костного мозга. Новые стволовые клетки могут выработать здоровые элементы кровеносной системы. Но операция сопряжена с рядом высоких рисков.
Терапия препаратами интерферона назначается обычно в 1-й стадии ХМЛ, так как не обладает эффективностью при последующих.
Для уменьшения массы опухоли и при отсутствии результата в лечении ингибиторами проводится химиотерапия. В стадии бластного криза используется полихимиотерапия аналогично лечению острого лейкоза.
Лучевая терапия может быть назначена в случае выраженной спленомегалии. При риске разрыва селезёнки проводят спленэктомию.
На сегодняшний день продолжаются исследования для создания ещё более совершенного препарата. Российскими учёными при помощи фонда «Сколково» проводятся клинические испытания ингибитора III поколения, который должен превзойти предыдущие по своей эффективности.
Профилактика и прогноз

Прогноз заболевания определяет врач
Причина образования ХМЛ не установлена, поэтому профилактикой являются меры по избеганию контактов с канцерогенными веществами, воздействия радиоактивного облучения.
Прогноз определяется стадией и тяжестью болезни. Одна из прогностических моделей (Kantarjian H.M.) включает факторы:
- преклонный возраст пациента при постановке диагноза;
- концентрация бластных клеток в крови ≥ 3%, в костном мозге ≥ 5%;
- концентрация базофилов ≥ 7%;
- концентрация тромбоцитов ≥ 700*10 9/л;
- выраженная спленомегалия.
Данная модель разработана для начальной фазы ХМЛ, если в наличии ≥ 3 признаков, прогноз неблагоприятный, последующие фазы рассматриваются как «всегда неблагоприятные». Однако каждый случай ХМЛ индивидуален, известны пациенты с продолжительностью жизни более 30 лет в хронической стадии. В среднем при своевременно начатом лечении ингибиторами тирозинкиназы 70-80% больных живут более 10 лет. При переходе болезни в прогрессирующую фазу выживаемость снижается в 3 — 4 раза, при бластном кризе по-прежнему составляет до 6 месяцев.